
Introduction
Neuroendocrine tumors (NETs) include a heterogeneous group of malignancies characterized by a relatively indolent rate of growth and a propensity to secrete a variety of hormones and biogenic amines. They arise from neuroendocrine cells, which are mainly located throughout the length of the gastroenteropancreatic (GEP) tract and the bronchopulmonary tree. The majority of NETs are sporadic, but they can also occur in the context of inherited familial syndromes, such as multiple endocrine neoplasia type 1 (MEN1), Von-Hippel Lindau syndrome, tuberous sclerosis and neurofibromatosis type 1, thus suggesting a causal role for genetic alterations during the tumorigenic process [1].
In recent years, a very heterogeneous picture of the genetic landscape of well-differentiated foregut, midgut and hindgut NETs has been depicted. Mutations of covalent histone modifiers including MEN1, PSIP1, SETD1B and members of the Polycomb complex have been observed in 40% of pulmonary carcinoids, and alterations in chromatin-remodeling genes have been described as sufficient to drive early steps in lung NET tumorigenesis [2]. In pancreatic NETs (pNETs), mutations of the epigenetic regulators MEN1 and DAXX/ATRX have been described in 44% and 43% of tumors respectively, while alterations of the mammalian target of rapamycin (mTOR) pathway have been found in 14% of the specimens [3]. Whole-genome and -exome sequencing has demonstrated that small bowel NETs are mutationally quiet, with a mutational burden of 0.1 somatic single nucleotide variants (SSNVs) per 105 nucleotides. Accordingly, recurrent mutations in the cyclin-dependent kinase inhibitor gene CDKN1B have been identified in only ~8% of tumors, in the absence of other obvious pathogenetic genomic alterations [4]. However, multiple epigenetic aberrations have been recently demonstrated in small bowel NETs, and their involvement in disease pathogenesis has been postulated [5]. Although patterns of gene mutations are highly diverse in NETs of different primary sites, classical oncogenes or tumor suppressors implicated in the development of many solid tumors (such as P53, RB or KRAS) do not appear to play a major role in the pathogenesis of any NETs [6]. In contrast, epigenetic dysregulation and/or alterations of the chromatin remodeling machinery seem to be a common element across different histologies. Thus, whether low-to-intermediate grade NETs are genetically-driven neoplasms or epigenetically-induced malignancies remains a legitimate though unanswered question.
Epigenetic modifications such as DNA methylation or histone acetylation, methylation and phosphorylation can cause heritable changes in gene expression without concomitant alterations in the genome of a cell. DNA methylation occurs primarily within the CpG islands located in the promoter regions and dictates the transcriptional potential of downstream target genes. Concomitantly, covalent histone modifications determine how DNA is packaged in nucleosomes, thus modulating the accessibility of underlying genes to transcription factors [7]. In addition, microRNAs (miRNAs), small single-stranded RNA molecules of ~19-22 nucleotides, regulate the gene expression at the post-transcriptional level, and have recently emerged as prominent epigenetic regulators [8]. Acting combinatorially, these mechanisms concur to determine the cellular phenotype, and there is increasing evidence that epigenetic aberrations are as relevant as gene mutations in the cancer pathogenesis (Figure 1) [9].
In this review, we provide an overview of the current knowledge on epigenetic changes in both hereditary and sporadic NETs of the GEP and bronchial tract, and focus on both diagnostic, prognostic and therapeutic implications that the NET epigenome profiling carries in the precision medicine era.
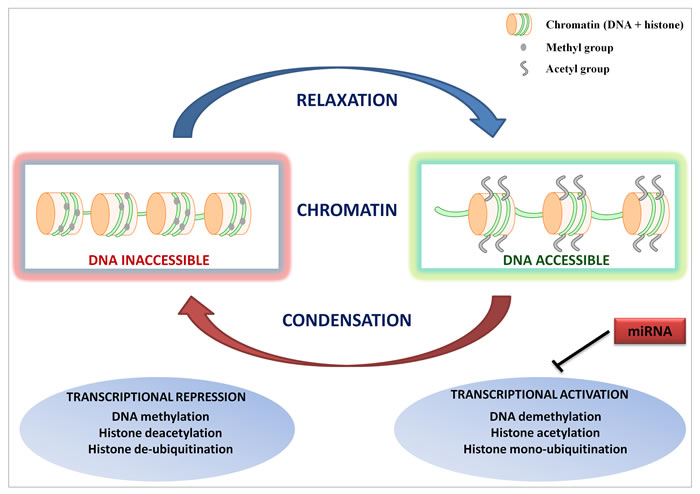
Figure 1: Epigenetic regulation of gene expression. Epigenetic alterations such as DNA methylation and/or histone modifications modulate the accessibility of genes to the transcriptional machinery by inducing either a relaxed/open or condensed/closed chromatin configuration. miRNAs concur to regulate the cell phenotype by repressing the expression of gene transcripts.
The epigenetic landscape of NETs
During the past decade, next-generation DNA and RNA sequencing methods have provided a more detailed picture of the epigenetic landscape of NETs. In particular, genome-wide approaches have contributed to define a molecular classification of these malignancies, thus paving the road for new diagnostic tools and innovative personalized therapies. Neuroendocrine lineage allocation is partly caused by progressive accumulation of complex layers of epigenetic modifications during the differentiation process from pluripotent endodermal cells [10, 11]. Although phenotypic stability of the differentiated cell state is secured by the so called “epigenetic memory” (reviewed in [12]), heterogeneous epigenetic profiles have been shown in NETs of different primary sites, thus suggesting underlying difference in the tumorigenic process, microenvironment-driven modulation of epigenetic states, and/or their possible correlation with the biological aggressiveness of these diverse neoplasms.
Familial neuroendocrine syndromes
Genetic and clinical features of familial neuroendocrine syndromes are summarized in Table 1. MEN1 is an autosomal-dominant syndrome characterized by tumors of the anterior pituitary, parathyroid glands and pancreaticoduodenal neuroendocrine cells, most commonly gastrinomas. It is caused by an inactivating mutation of the MEN1 gene, which encodes for menin, a nuclear protein implicated in cell division, genome stability, and transcription regulation via histone methylation. Up to 10% of patients with MEN1 syndrome may not harbor mutations in the coding regions of the MEN1 gene, but in the gene promoter or untranslated regions, challenging the genetic diagnosis [13]. As a constituent of a multiple protein complex displaying a histone H3 lysine 4 methyltransferase activity, MEN1 has a critical role in chromatin remodeling. In particular, MEN1 acts as either repressor or activator of gene transcription through interaction with a plethora of histone deacethylases (HDACs) and histone methyltransferases including PRMT5 and SUV39H1. Epigenetic silencing of the Hedgehog pathway, of the homeobox gene GBX2 as well as of the gastrin-encoding gene GAST has been reported downstream of MEN1 [14-16]. On the other hand, transcriptional activation of the HOX cluster (HOXA9, HOXC6, and HOXC8) and cyclin-dependent kinase inhibitor (CDKN1B, CDKN2C) genes has been documented, but the biologic consequences on pNET tumorigenesis need to be clarified [17-19]. To elucidate the genome-wide transcriptional modifications induced by MEN1 through epigenetic remodeling in pancreatic islets, a recent study integrated gene expression profile analysis and histone H3 lysine 4 trimethylation (H3K4me3) mapping, and identified insulin-like growth factor 2 mRNA binding protein 2 (IGF2BP2) gene as a target subjected to MEN1 dynamic regulation. IGF2BP2 interferes with IGF2 translation during the embryonic development, and its dysregulation upon MEN1 loss might play a role in pNET pathogenesis [20].
In neurofibromatosis type 1 and tuberous sclerosis syndrome, epigenetic silencing of the wild-type NF1 or TSC2 genes has been proposed as a possible tumorigenic event, in accordance with the Knudson’s two-hit hypothesis [21, 22]. Although data in lung and GEP-NETs arising in the context of Von-Hippel Lindau syndrome (VHL) are lacking, there is evidence that mutations of multiple chromatin remodelers including the histone methyltransferase SETD2 and the histone demethylases UTX and JARID1C may contribute to the progression of VHL-associated clear renal cell carcinoma [23].
Table 1: Familial neuroendocrine syndromes: genetic and clinical features.
Syndrome |
Causative gene |
Gene location |
Protein |
GEP-NET type (penetrance) |
MEN1 |
MEN1 |
11q13 |
Menin |
Gastrinoma (40%) Non-functioning pNET (20%) Insulinoma (10%) Glucagonoma <1% VIPoma <1% Gastric carcinoid 10% |
VHL syndrome |
VHL |
3p25 |
VHL |
Non-functioning pNET (12-17%) |
Tuberous sclerosis |
TSC1/TSC2 |
9q34/16p13 |
Hamartin/tuberin |
pNET (<5%) |
NF1 |
NF1 |
17q11.2 |
Neurofibromin |
Somatostatinoma (6%) |
Pancreatic NETs
A number of studies have investigated the epigenetic changes possibly related to pNET pathogenesis and progression, and hypermethylation of the promoters of RASSF1, CDKN2A, TIMP3, MGMT, MLH1 and IGF2 genes has been reported (Table 2). Ras association domain family 1 (RASSF1) is a tumor suppressor gene consisting of eight exons alternatively spliced to encode 8 protein isoforms, RASSF1A-H. RASSF1A is involved in microtubule stabilization, cell cycle regulation and induction of apoptosis [24], and the aberrant methylation of its promoter has been observed in 60-100% of pNETs [25-29]. Of interest, the transcription levels of RASSF1A are inversely correlated with the degree of gene methylation [30], and RASSF1A hypermethylation seems to predict pNET malignant features such as larger tumor diameter, nodal involvement and hepatic metastases [26, 28]. Cyclin-dependent kinase inhibitor 2A (CDKN2A) encodes for the tumor suppressor protein p16, which concurs to regulate cell cycle progression by inhibition of the G1/S transition. In a study of 48 well-differentiated pNETs, hypermethylation of CDKN2A was observed in 40% of tumors and was significantly associated with decreased patient survival and early tumor recurrence after surgery [26]. Of note, CDKN2A hypermethylation seems to be a hallmark of gastrinomas, since it occurs in 52-62% of gastrinomas but only in 17% of insulinomas [31-33]. Loss of p16 as result of gene promoter methylation is not associated with disease stage or prognosis, thus suggesting its early occurrence in gastrinoma pathogenesis [31, 32]. In vitro, reacquisition of p16 expression after treatment with the hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) resulted in growth inhibition of pNET cells [34]. Methylation of the tumor suppressor tissue inhibitor of metalloproteinase-3 (TIMP3) has been found in 8/18 (44%) samples of pNETs, and has been correlated with loss or reduction of protein expression. TIMP3-negative tumors are at increased risk of lymph node and/or liver metastases, whereas they are apparently never associated with ectopic insulin production [35]. The suicide enzyme O6-methylguanine DNA methyltransferase (MGMT) repairs DNA by removing the O6-alkylguanine adducts, and its role in pNET chemoresistance to alkylating agents including temozolomide has been widely investigated, with controversial results [36-39]. Methylation in the promoter region of MGMT has been observed in up to 56% of pNETs, and only a partial concordance with protein expression has been demonstrated, thus suggesting the existence of various mechanisms of MGMT expression regulation in addition to transcriptional modulation [38]. Both MutL homolog 1 (MLH1) and Insulin-like growth factor 2 (IGF2) are frequently methylated in insulinomas, but not in other pNETs [40, 41]. Accordingly, in a recent study investigating the DNA methylation level of 807 cancer-related genes in insulinomas, gastrinomas and non-functioning pNETs, DNA methylation patterns were found to be specific for each tumor subtype [42]. An overview of recurring epigenomic difference between insulinomas and other pNETs is provided in Figure 2. Along with fundamental difference in the genetic background [3, 43], epigenetic changes peculiar of insulinomas might contribute to determine their unique clinical behavior.
Table 2: Incomplete list of epigenetic changes in sporadic NETs by primary site.
NET primary site |
Epigenetic alteration |
Reference |
Pancreas |
Promoter hypermethylation: RASSF1A CDKN2A TIMP3 MGMT MLH1 IGF2 Axin-2, SFRPs CIMP positivity Histone modifications: Upregulation of histone H3K9me2 miRNA upregulation: miR-103, -107, -23a, -26b, -192, -342 miR-144/451 miRNA downregulation: miR-155 IncRNA downregulation: MEG3 |
[37] [40] [42] [43] [46] [31] [46] [51] [52] [51] [53] |
Small bowel |
Promoter hypermethylation: RASSF1A TCEB3C THBS1, MGMT, p14, p16 LAMA3, SERPINB5, RANK Global hypomethylation Histone modifications: Upregulation of histone H1x Upregulation of H3K4diMe miRNA upregulation: miR-183, -488, - 19a+b miR-96, -182, -183, - 196a, 200a miRNA downregulation: miR-133a, -145, -146, -222, -10b miR-31, -129-5p, 133a, -215 |
[54] [58] [59] [5] [64] [65] [66] [67] [66] [67] |
Lung |
Promoter hypermethylation: RASSF1A P15INK4b Histone modifications: Downregulation of H4KM20 and H4KA16 miRNA upregulation: miR-129, -323-3p, -487b, -410, -369-3p, 376a miRNA downregulation: miR-203, -224, -155, -302, -34b, -181b, -193a, -5p, -34b |
[69] [70] [73] [74] [74] |
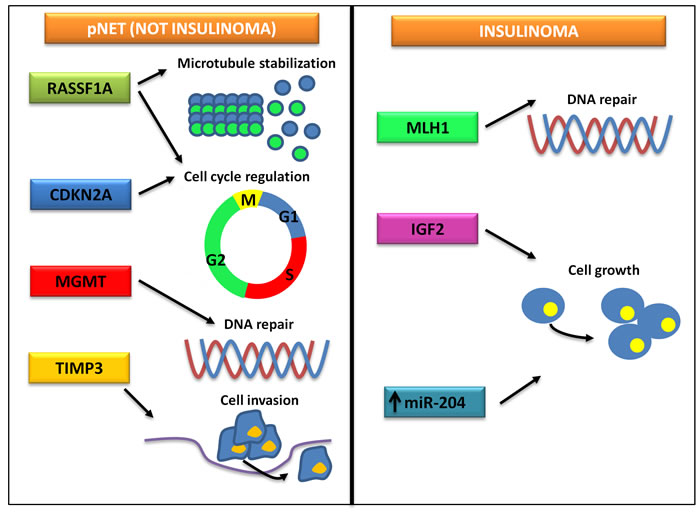
Figure 2: Frequent epigenetic modifications in insulinomas and other pNETs. While the epigenetic landscape of insulinomas is characterized by alterations of the signaling of MLH1 and IGF2, non-insulinoma pNETs are defined by a different pattern of epigenetic changes, eventually leading to cell cycle dysregulation, increased cell motility and chemoresistance.
Hyperactivation of the Wnt/β-catenin signaling contributes to the pathogenesis and progression of pNETs. Although mutations of β-catenin or Wnt antagonists such as APC are rare, epigenetic silencing of Wnt inhibitors including Axin-2, secreted Frizzled-related proteins (SFRPs), Wnt inhibitory factor-1 (WIF-1) and DICKKOPFs (DKKs) has been reported in pNET cell lines. In particular, while silencing of SFRPs and Axin-2 was related to the promoter methylation, downregulation of WIF-1, DKK-1 and DKK-3 was caused by repressive histone modifications leading to increased H3K9me2 presence at promoter level. Interestingly, treatment with decitabine was able to restore the expression of these genes, resulting in tumor suppressor functions both in vitro and in vivo [44].
Tumors that are characterized by frequent promoter methylation of tumor suppressor genes harbor the so called CpG island methylator phenotype (CIMP). CIMP positivity has been observed in 83% of pNETs versus 66% of extra-pancreatic NETs, and is apparently correlated with high proliferation index (Ki-67 > 10%) and poor prognosis, at least based on descriptive survival analyses [29]. In a study of 56 low-to-intermediate grade pNETs, methylation-sensitive multiple ligation-dependent probe amplification and long-interspersed nucleotide element-1 (LINE-1) analysis were used to assess tumor hypermethylation and hypomethylation status respectively. Futhermore, unsupervised hierarchical clustering allowed to separate a group of pNETs lacking both hyper- and hypomethylation features, a group characterized by moderate promoter hypermethylation as well as LINE-1 hypomethylation, and a highly hypermethylated group. The highly hypermethylated tumor phenotype predicts unfavorable disease progression, and is apparently associated with stage IV [29, 45].
Recently, a genome-wide DNA methylation study has been performed to assess the epigenetic consequences of DAXX/ATRX loss in pNETs. Alterations in the DNA methylation profile were more profound in DAXX-negative pNETs rather than in ATRX-negative or DAXX/ATRX-positive tumors, suggesting that the deficiency of DAXX, but not ATRX, drives genome-wide methylation changes leading to pNET formation [46]. DAXX and ATRX exert multiple functions including chromatin remodeling during heterochromatin assembly at repetitive guanine-rich regions, where they are required for incorporation of the histone variant H3.3 [47]. Mutations of DAXX or ATRX are mutually exclusive, and result in nuclear protein loss with consequent activation of the alternative lengthening of telomeres (ALT) pathway, a mechanism of telomerase-independent telomere maintenance. This eventually leads to chromosomal instability (CIN), tumor heterogeneity and metastases [48]. Histone modifications have been poorly studied in pNETs. However, the recurrent mutations of MEN1 in sporadic pNETs suggest covalent histone changes as possible drivers of disease pathogenesis.
Studies of miRNAs in pNETs are scarse. In a series of pancreatic cancers and normal tissue, overexpression of miR-103 and miR-107 with concurrent miR-155 downregulation was distinctive of tumors, but did not separate pNETs from pancreatic acinar carcinomas. As compared with non-functioning pNETs, insulinomas were enriched in miR-204 [49]. In another study, a peculiar miRNA expression profile was shown to distinguish between insulinomas and normal pancreatic islets, and the miRNA cluster miR-144/451 was found to promote NET cell proliferation through suppression of the PTEN/AKT pathway [50]. Long non-coding RNAs (IncRNAs) have been implicated in pNET pathogenesis downstream of MEN1 biallelic inactivation. In particular, the IncRNA maternally expressed gene 3 (MEG3) is downregulated in pNETs, thus leading to the overexpression of its target protooncogene c-Met and consequently increased cell proliferation [51].
Small bowel NETs
Similarly to pNETs, downregulation of RASSF1A as result of gene promoter methylation has been implicated in the progression of small bowel carcinoid tumors. In a study of 33 small bowel NETs and matched metastases, methylation levels of RASSF1A and CTNNB1, the gene encoding β-catenin, were significantly higher in metastatic tissue as compared with primaries. Of interest, both genes were unmethylated in an additional cohort of 6 nonmetastatic appendiceal carcinoids, thus suggesting that methylation of these genes is required for metastasis development [52]. More recently, increased methylation and consequent transcriptional repression of RASSF1A, but not CTNNB1, has been shown in metastases rather than small bowel primary NETs. Notably, low expression of RASSF1A significantly predicted poor survival and was associated with loss of chromosome 16q. In vitro, treatment with decitabine resulted in restored expression of RASSF1A in NET cell lines [53].
DNA methyltransferases (DNMTs) are the enzymes primarily responsible for DNA methylation. In particular, while DNMT1 maintains genome methylation during cellular replication, DNMT3a and DNMT3b act as de novo methyltransferases [54]. In a series of 63 foregut, midgut and hindgut GEP-NETs, DNMT1, 3a and 3b were expressed in 87%, 81% and 75% of samples respectively, and their expression was significantly higher in stage IV tumors. Of interest, DNMT3a and 3b were significantly down-regulated in midgut tumors relative to the foregut or hindgut NETs [55], thus suggesting that distinct epigenetic alterations characterize these diverse entities. Transcription Elongation Factor B Polypeptide 3C (TCEB3C) encodes for Elongin A3, which interacts with Elongin BC thus increasing the RNA polymerase II transcription elongation potential. TCEB3C is the only known imprinted gene on chromosome 18, and frequent loss of chromosome 18 has been reported in small bowel NETs. Epigenetic repression of TCEB3C has been recently observed in midgut carcinoids, and is apparently related to both DNA and histone methylation. Both decitabine, a demethylating agent, and the histone methyltransferase inhibitor 3-deazaneplanocin A and DNMT1 silencing via siRNA induced TCEB3C expression, thus leading to decreased carcinoid cell survival [56]. Using a candidate gene-driven approach, Chan et al. demonstrated that p14, p16, thrombospondin 1 (THBS1) and MGMT were selectively hypermethylated in midgut carcinoids when compared with the adjoining normal mucosa. CpG island methylation of p16 also correlated with patient age and metastatic status [57]. CIMP positivity has been demonstrated in up to 29% of well-differentiated foregut and midgut NETs, and did not affect patient survival [29].
Although its importance is often underestimated as compared with the hypermethylation of the promoter of tumor-suppressor genes, global hypomethylation is a key epigenetic feature of human tumors. It refers to the methylation levels of DNA repeat elements such as short interspersed (Alu) and long interspersed nucleotide elements (LINE-1), which constitute approximately half of the genome, and is correlated with chromosomal instability and increased tumor mutation rate [58]. Contrasting results have been reported so far regarding the global methylation pattern of small bowel carcinoids. By LINE-1 and Alu methylation analyses, intestinal NETs were shown to be hypomethylated as compared with normal tissue, and this feature was more prevalent in ileal carcinoids rather than in other NETs, and was associated with lymph node metastases [59]. However, in a subsequent study, hypomethylation was more marked in pNETs, while NETs metastatic to lymph nodes were less frequently hypomethylated than nonmetastatic tumors [60]. More recently, Fotouhi et al. observed significantly lower methylation levels in small bowel carcinoids when compared to normal ileum, as well as in distant metastases rather than in primary tumors. Global hypomethylation was also associated with chromosome 18 loss [53]. In an attempt to define the epigenetic changes that take place during metastatic progression, the methylation profile of 10 small intestine NETs and 10 matched lymph node metastases has been recently compared. Overall, hypomethylation was more pronounced in metastatic tissue, and a large number of genes regulating cell growth, apoptosis, proliferation and metastasis formation were found to be differentially methylated between primaries and metastases. However, definite conclusions were hindered by the small sample size of the study [61].
In a large-scale multilocus analysis of DNA methylation patterns, two distinct groups of small intestine NETs were identified based on their DNA global methylation profile. Of interest, these two groups did not show any histological differences, including proliferation index, and were not associated with other known prognostic factors, thus suggesting possible pathogenetic differences lacking relevant consequences on tumor behavior [42]. Recently, Karpathakis et al. [5] have characterized 97 small bowel NETs by genetic, epigenetic and transcriptional profiling, and identified three subgroups of tumors by integrated molecular analysis. The largest group of NETs was defined by chromosome 18 loss of heterozygosity, and was associated with CDKN1B mutations and CIMP negativity. Patients of this subgroup had the most favorable progression-free survival (PFS) after resection and presented at an older age, thus suggesting a less aggressive tumor phenotype. On the other hand, a second subgroup was characterized by the absence of arm-level copy number variations (CNVs), a high level of CIMP and an intermediate PFS, while the third subgroup was defined by multiple CNVs, younger age at onset and dismal PFS. Overall, up to 85% of small bowel carcinoids harbored epimutations, and 21 epigenetically silenced genes were identified. Among those, there were genes located on chromosome 18 such as laminin alpha 3 (LAMA3), serpin peptidase inhibitor clade B member 5 (SERPINB5) and receptor activator of nuclear factor-κB (RANK). Because of the frequent LOH of chromosome 18 in small intestine NETs, epigenetic dysregulation of these genes has been proposed as the “second hit” capable to drive the tumorigenic process.
Studies on histone modifications in small bowel NETs are limited. Overexpression of histone variant H1x in gastrointestinal NETs seems to reflect its abundance in normal neuroendocrine cells, and might be useful for differential diagnosis in equivocal cases [62]. In 16 small intestine NETs, high levels of dimethylation of histone H3 at lysine 4 (H3K4diMe) have been observed, but further studies are needed to define the molecular implications of this finding in NET pathogenesis [63].
Small bowel NET progression is apparently characterized by a differential pattern of miRNA expression. In a study of 8 ileal carcinoid tumors, miR-183, -488, and 19a+b were up-regulated while miR-133a, -145, 146, -222 and -10b were down-regulated in metastatic tissue with respect to primary tumors [64]. In a subsequent study, 24 small intestinal NETs at different stages were profiled in their miRNA expression, and up-regulation of miR-96, -182, -183, -196a and 200a as well as down-regulation of miR-31, -129-5p, -133a and -215 were reported during tumor progression [65]. Up-regulation of miR-183 and down-regulation of miR-133a in both studies makes these miRNAs appealing targets for future investigations. Moreover, recent evidence suggests that miR-129-5p may have an anti-proliferative and anti-metastatic effect in midgut carcinoid tumors, and that its down-regulation during tumor progression might affect factors involved in RNA binding and nucleotide metabolism such as EGR1 and G3BP1 [66].
Lung NETs
Chromatin remodeling and epigenetic dysregulation have been depicted as key events in the pathogenesis of pulmonary carcinoids. A recently published whole-genome/exome sequencing study of 69 lung NETs demonstrated mutually exclusive mutations of histone covalent modifiers in 40% of the samples. Among these genes, there were the menin-binding protein PSIP1, the histone methyltransferases SETD1B, SETDB1 and NSD1, the histone demethylases KDM4A, PHF8 and JMJD1C, as well as members of the Polycomb repressive histone H3K27 methyltransferase complexes CBX6, EZH1 and YY1. Numerous mutations were also found in ATP-dependent chromatin remodeling SWI/SNF complex members including ARID1, ARID2, SMARCA1, SMARCA2, SMARCA4,SMARCC2, SMARCB1 and BCL11A. Given the almost universal absence of other cancer-related mutations, alteration of chromatin-remodeling pathways is apparently sufficient to drive early steps of lung NET tumorigenesis [2].
As previously reported for pNETs and small bowel carcinoids, RASSF1 promoter hypermethylation is a frequent event in low-to intermediate grade lung NETs. Of interest, the degree of promoter methylation was associated with tumor grade, while there was a non-linear correlation between levels of methylation and RASSF1 mRNA or protein content, thus suggesting that other transcriptional or post-transcriptional events may concur in its regulation [67]. In a series of 5 low-grade and 15 high-grade lung NETs, aberrant methylation at the 5’-region of the p15INK4b gene was observed in 15% of tumors, but not in normal bronchial tissue. p15INK4b encodes for a cyclin-dependent kinase inhibitor, and has recently emerged as a possible regulator of tumorigenesis via cell cycle dysregulation [68]. The protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme, and is overexpressed in bronchial NETs. In particular, nuclear overexpression of PRMT5 was negatively associated with tumor grade, thus reflecting potential differences in the epigenetic control of oncogenesis of low- and high-grade lung NETs [69]. Consistently with this hypothesis, expression levels of the histone methyltransferase enhancer of zeste homolog 2 (EZH2) have been shown to inversely correlate with tumor grade in lung NETs [70].
Comprehensive analyses of histone modifications in bronchial carcinoids are lacking. In a study of 32 lung NETs, histone H4 acetylation at lysine 16 (H4KA16) and trimethylation at lysine 20 (H4KM20) were studied. As in other cancers, a progressive loss of H4KM20 and H4KA16 was observed during progression from normal bronchiolar epithelium to low grade tumors and then to high-grade carcinomas. The biological implications underlying this finding need to be clarified [71].
Several studies have recently profiled the expression of miRNAs in pulmonary carcinoids, reporting differences between normal lung tissue and tumor, low- and high-grade bronchial NETs as well as localized and metastatic disease [72-75]. Among recurrently deregulated miRNAs in pulmonary carcinoids, miR-21, miR-155 and miR-129-5p were already identified as possible regulators of pNET or midgut NET pathogenesis, thus suggesting their pivotal role in NET oncogenesis and progression.
Clinical oncoepigenomics of NETs in the precision medicine era
Precision medicine refers to individualized prevention and treatment of diseases based on their underlying molecular causes, and carries the promise of coupling established clinical-pathological parameters with state-of-the-art molecular profiling to create diagnostic, prognostic and therapeutic strategies tailored to each patient’s requirements. Data from the Human Genome Project and the global diffusion of next-generation sequencing technologies have dramatically advanced the practice of personalized medicine, and many cancers are currently treated using agents targeting underlying aberrant genomic changes [76].
The genomic landscape of NETs has been decoded only very recently, and the clinical relevance of peculiar mutational profiles is still under investigation. However, no genetic signature can be used for treatment tailoring in NET patients at present, even when driver mutations affect the signaling pathways targeted by currently approved drugs. While genetic features of different site NETs are extremely heterogeneous, a limited number of pathways seem to have a role in their pathogenesis, as demonstrated by the almost universal efficacy of somatostatin analogs and mTOR inhibition in these tumors [77, 78]. Since disruption of the epigenetic machinery is common to all NETs, a possible molecular reunification of such a diverse disease can be envisaged, thus explaining their similar response to several therapeutic agents. Although the NET epigenome characterization is still in its infancy, epigenetic profiles have already demonstrated clinical utility for the diagnosis and prognosis of NETs, and might pave the way to new personalized therapeutic strategies.
Epigenetic profiles in NET diagnosis and prognosis
A whole-genome DNA methylation analysis of 12 normal lung tissue samples and 124 lung tumors has recently identified five DNA methylation subgroups. Of note, one epitype was distinctly associated with neuroendocrine differentiation [79]. Similarly, small bowel carcinoids and distinct pNET subtypes have been shown to harbor peculiar DNA methylation patterns [42]. Discrimination of the various subtypes of pulmonary NETs via miRNA profiling has been demonstrated to be feasible [80]. Recently, an integrated multi-omics data analysis has assessed the transcriptomic (mRNA and miRNA), mutatomic (selected mutations) and metabolomic profile of pNETs, identifying three distinctive molecular subtypes. While the islet/insulinoma tumor (IT) subtype was characterized by low grade and low metastatic potential, the metastasis-like primary (MLP) subtype displayed high proliferative activity, a clinically aggressive behavior and a gene expression profile consistent with epithelial-to-mesenchymal transition (EMT). On the other hand, the MEN1-like/intermediate subtype was enriched in MEN1 mutations, and showed moderate metastatic potential. Although validation studies are needed, this seminal molecular classification of pNETs shows promising clinical applicability, potentially leading to a better therapeutic planning [81]. Observations from our group are also in line with this classification. In fact, we have previously reported that osteotropic NETs overactivate EMT, while being characterized by very dismal prognosis [82]. Given the apparent pivotal role of CXCR4 in inducing EMT and metastases in NETs (manuscript submitted), and the robust epigenetic regulation of the CXCL12/CXCR4 axis in cancer [83], future work should determine if complex NET epigenetic profiles and resulting patterns of biologic malignancy as well as clinical outcomes may be recapitulated, at least in part, by CXCR4 expression.
A multianalyte whole blood RNA multigene signature has been developed to predict NET activity. Interestingly, among the omic clusters capable of differentiating disease activity (stable versus progressive disease), there was the epigenome [84]. Future studies should assess if the epigenomic profiling per se may serve as a predictive biomarker for treatment tailoring in NET patients. In this context, the epigenetic silencing of MGMT has been described as predictive of response to alkylating agents in pNETs [38, 39], but prospective validation is still lacking. The prospective randomized trial NCT01824875, investigating temozolomide alone or in combination with capecitabine in pNET patients, incorporates MGMT promoter methylation testing, and may therefore provide further insight on this topic.
Epigenetic clustering has shown prognostic relevance in GEP-NETs, while studies in bronchial carcinoids are lacking. In low-to-intermediate pNETs, the combination of DNA global hypomethylation and cluster gene hypermethylation significantly predicted patient outcomes in multivariable analysis [45]. In addition, high expression of miR-196a is apparently associated with decreased overall survival (OS) and disease-free survival (DFS) in pNET patients [85]. As described above, the CIMP status has demonstrated prognostic relevance in small bowel NETs, with a substantial impact on patient PFS [5].
Epigenetic therapy of NETs
Since alterations of the chromatin remodeling machinery seem to be major drivers of NET development, epigenetic agents such as DNMT antagonists or HDAC inhibitors may be effective for patients with NETs. Moreover, in contrast to DNA mutations, DNA methylation and histone modifications are reversible and seem to be a feature defining cancer stem cells [86], thus representing an appealing therapeutic target in cancer. An overview of the epigenetic agents currently approved by FDA for cancer patients, their mechanism of action and clinical indications is provided in Table 3.
Table 3: Approved epigenetic agents in the treatment of cancer in Europe and North America.
Drug |
Mechanism of action |
Approved indications |
First FDA approval |
Azacitidine |
Inhibition of DNA methyltransferases |
Myelodysplastic syndromes (FDA, EMA) Acute myeloid leukemia (EMA) Chronic myelomonocytic leukemia (EMA) |
2004 |
Decitabine |
Inhibition of DNA methyltransferases |
Myelodysplastic syndromes (FDA) Acute myeloid leukemia (EMA) |
2006 |
Vorinostat |
Inhibition of histone deacetylases (class I and II) |
Cutaneous T-cell lymphoma (FDA, EMA) |
2006 |
Romidepsin |
Inhibition of histone deacetylases (1,2,4,6) |
Cutaneous T-cell lymphoma (FDA) |
2009 |
Belinostat |
Inhibition of histone deacetylases (class I, II and IV) |
Peripheral T-cell lymphoma (FDA) |
2014 |
Panobinostat |
Inhibition of histone deacetylases (class I and II) |
Multiple myeloma (FDA, EMA) |
2015 |
The efficacy of DNMT inhibitors in NETs has been tested only in vitro thus far. Azacytidine caused a dose-dependent reduction of the proliferation of CNDT2.5, H727 and BON1 cell lines [87]. Similarly, the demethylating agent decitabine showed antiproliferative effects on QGP1 pNET cells, possibly as result of the restoration of multiple genes silenced by pathogenetic de novo methylation. In particular, after treatment with the drug, tumor cells expressed a differential pattern of genes involved in proliferation, apoptosis and metastases [34]. Decitabine was also able to restore the expression of RASSF1A in bronchial NET cell lines [52].
HDAC inhibitors such as trichostatin A, sodium butyrate and entinostat have been tested on pNET cell lines, and caused a dose-dependent inhibition of proliferation, cell cycle arrest and apoptosis. No synergistic effects were noted after combination with somatostatin or octreotide [88]. The antiproliferative effects of valproic acid (VPA) as a class I and IIa HDAC inhibitor have been extensively investigated in NETs. The drug is able to induce a dose-dependent growth inhibition of NET cells of pancreatic and intestinal origin in vitro through activation of both intrinsic and extrinsic apoptosis. Of note, expression of up to 20% of protein-coding genes was significantly modified by VPA in NET cell lines, leading to major alterations in key regulatory pathways including the signaling of p53, TGFβ1 and MYC [89]. VPA has also been demonstrated to up-regulate Notch1 and somatostatin receptor subtype 2 (SSTR2). While activation of Notch1 is able per se to suppress NET tumor growth and is associated with reduction of NET tumor markers [90, 91], SSTR2 up-regulation has been used to synergistically increase the cytotoxicity of a SSTR-targeting camptothecin-somatostatin conjugate [92]. Concurrent activation of the Notch1 pathway and inhibition of the GSK3β signaling by combination of VPA and lithium has been reported to suppress NET cell proliferation, while decreasing CgA production [93].
Early evidence showed that VPA exerts antisecretory effects in NETs, significantly decreasing the plasma concentration of somatostatin in a single patient with somatostatinoma [94]. In a pilot phase II study of eight patients with low-grade pNETs or midgut carcinoids, VPA at 500 mg daily induced an unconfirmed partial response and disease stabilization in one and four subjects respectively. Of note, Notch1 upregulation following treatment was apparently associated with better outcomes [95]. The HDAC inhibitor depsipeptide has been investigated in a phase II study of 15 patients with metastatic NETs. The trial has been prematurely discontinued due to an unexpected rate of severe cardiac toxicities, including a fatal ventricular arrhythmia [96]. In a single arm phase II study of 15 patients with GEP-NETs, panobinostat was associated with a 90% rate of disease stabilization and a median PFS of 11.8 months. The trial was stopped at interim analysis due to lack of objective responses [97]. However, it should be noted that response rate is currently not considered an optimal endpoint for NET clinical trials [98]. The DNA methylation and deacetylation inhibitor RRx-001 has recently demonstrated to inhibit hormone release in a single patient with carcinoid syndrome, leading to rapid symptom reversal and improved quality of life [99]. However, future studies are needed to confirm this observation.
Modulators of H3K4me3 demethylases of the KDM5 family are currently being developed, and have already shown significant antitumor activity in murine models of MEN1 knock-out pNETs [20]. Inhibition of KDM5 proteins could be an attractive future strategy in NET patients, and MEN1 deficiency might be envisaged as a potential predictor of response. Although preclinical evidence of efficacy for epigenetic agents in NETs is promising, no significant benefit has been demonstrated so far by clinical trials. However, as a possible explanation for this lack of efficacy, it should be noted that these studies were carried out using HDACi, while the preclinical investigations of epigenetic agents in NETs have prevalently used demethylating agents. The only epigenetic modulator still under clinical investigation is belinostat, which is currently being trialled in combination with cisplatin and etoposide, in patients with advanced neuroendocrine carcinomas (https://clinicaltrials.gov/ct2/show/study/NCT00926640?term = nct00926640&rank = 1).
Conclusions and future perspectives
Since commonly mutated oncogenes play little or no pathogenetic role in NETs, epigenetic alterations are likely to be major determinants of NET tumorigenesis. However, whether the observed modifications of DNA packaging are driver or passenger events, and their position and role in the evolutionary tree of NETs still remains to be clarified. Recently, several studies have compared the genomic make-up of NET cell lines to the genetic signature of primary NETs, demonstrating striking differences in the rates and patterns of mutations [100, 101]. Future work is needed to assess the degree of overlap between the epigenetic landscape of NET cell lines and correspondent patient tumors. In the meanwhile, the results from experiments with NET cell lines should be interpreted with caution, and direct clinical extrapolation should be avoided.
Although profiles of the epigenome of NETs have been already proposed, results need to be validated in larger studies. The identification of molecularly different NET subtypes might have a dramatic impact on clinical practice, potentially leading to new diagnostic classification, prognostic stratification and innovative clinical trials. While precision medicine has thus far led to the “explosion” of each form of cancer in multiple distinct diseases, the common epigenetic changes seen in a wide spectrum of NETs have the potential of re-unifying such diverse clinical entities. Future investigations should verify if the epigenetic fingerprinting of NETs might provide better clinical classification/prognostic stratification than site of disease or grade. No biomarker-driven clinical trials of epidrugs have been carried out in NETs so far, thus possibly explaining the observed lack of significant efficacy with these agents. Similarly to studies of therapeutics targeting discrete genomic mutations, innovative basket trials testing epigenetic drugs should ideally include molecularly defined subpopulations of NET patients, thus paving the way to studies dedicated to emerging homogeneous epitypes of NET patients.
Since NETs originate from neuroendocrine cells, studies of epigenetic profiling should be carried out with appropriate control tissue. This might be a potential drawback for a precise characterization of small bowel or bronchial carcinoids, but innovative methods for single-cell genome-wide epigenomic analyses are now on the horizon [102]. Recent evidence [103, 104] suggests that epidrugs stimulate the expression of benign retroviruses inserted in the genome of all human cells, including tumor cells. As result, a “viral mimicry” phenomenon and its beneficial effects on tumor immunogenicity have been described, thus providing a rationale for possible combination studies in NETs with epidrugs and immune checkpoint inhibitors such as nivolumab or ipilimumab [105]. Impressive progress has been made in our understanding of NET pathogenesis in recent years, and the recognition of pivotal pathways of tumorigenesis has led to the approval of new targeted therapies for NET patients. Outstanding bench to bedside and back work is now again needed to further move from NET treatment to NET cure.
Acknowledgments
This work was supported by a grant from the Italian Association for Cancer Research (AIRC, IG17536) and by the “Oncogenomic Project” from the Apulia Region, 2015.
Conflicts of interest
The Authors declare no affiliation with industries or organizations with a financial interest, direct or indirect, that may affect the conduct or reporting of the work submitted.
References
1. Cives M, Strosberg J. An update on gastroenteropancreatic neuroendocrine tumors. Oncology (Williston Park). 2014; 28: 749-756.
2. Fernandez-Cuesta L, Peifer M, Lu X, Sun R, Ozreti L, Seidel D, Zander T, Leenders F, George J, Müller C, Dahmen I, Pinther B, Bosco G, et al. Frequent mutations in chromatin- remodelling genes in pulmonary carcinoids. Nat Commun. 2014; 5: 3518. doi: 10.1038/ncomms4518.
3. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, Velculescu VE, Diaz LA Jr, Vogelstein B, Kinzler KW, Hruban RH, Papadopoulos N. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011; 331: 1199-203. doi: 10.1126/science.1200609.
4. Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, Banck MS, Kanwar R, Kulkarni AA, Karpathakis A, Manzo V, Contractor T, Philips J, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013; 45: 1483-6. doi: 10.1038/ng.2821.
5. Karpathakis A, Dibra H, Pipinikas C, Feber A, Morris T, Francis J, Oukrif D, Mandair D, Pericleous M, Mohmaduvesh M, Serra S, Ogunbiyi O, Novelli M, et al. Prognostic Impact of Novel Molecular Subtypes of Small Intestinal Neuroendocrine Tumor. Clin Cancer Res. 2016; 22: 250-8. doi: 10.1158/1078-0432.CCR-15-0373.
6. Karpathakis A, Dibra H, Thirlwell C. Neuroendocrine tumours: cracking the epigenetic code. Endocr Relat Cancer. 2013; 20: R65-82. doi: 10.1530/ERC-12-0338.
7. Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015; 16: 519-32. doi: 10.1038/nrm4043.
8. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015; 15: 321-33. doi: 10.1038/nrc3932.
9. Graham JS, Kaye SB, Brown R. The promises and pitfalls of epigenetic therapies in solid tumours. Eur J Cancer. 2009; 45: 1129-36. doi: 10.1016/j.ejca.2009.01.003.
10. Rosai J. The origin of neuroendocrine tumors and the neural crest saga. Mod Pathol. 2011; 24 Suppl 2: S53-57. doi: 10.1038/modpathol.2010.166.
11. Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, Yang H, Wang T, Lee AY, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013; 153: 1134-48. doi: 10.1016/j.cell.2013.04.022.
12. D’Urso A, Brickner JH. Mechanisms of epigenetic memory. Trends Genet. 2014; 30: 230-6. doi: 10.1016/j.tig.2014.04.004.
13. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. 2014; 386: 2-15. doi: 10.1016/j.mce.2013.08.002.
14. Yang YJ, Song TY, Park J, Lee J, Lim J, Jang H, Kim YN, Yang JH, Song Y, Choi A, Lee HY, Jo CH, Han JW, Kim ST, Youn HD, Cho EJ. Menin mediates epigenetic regulation via histone H3 lysine 9 methylation. Cell Death Dis. 2013; 4: e583. doi: 10.1038/cddis.2013.98.
15. Gurung B, Feng Z, Iwamoto DV, Thiel A, Jin G, Fan CM, Ng JM, Curran T, Hua X. Menin epigenetically represses Hedgehog signaling in MEN1 tumor syndrome. Cancer Res. 2013; 73: 2650-8. doi: 10.1158/0008-5472.CAN-12-3158.
16. Kim H, Lee JE, Cho EJ, Liu JO, Youn HD. Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003; 63: 6135-9.
17. Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004; 13: 587-97.
18. Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004; 24: 5639-49. doi: 10.1128/MCB.24.13.5639-5649.2004.
19. Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt- Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, Hua X, Roeder RG, Meyerson M, Hess JL. Menin and MLL cooperatively regulate expression of cyclin- dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005; 102:749-54. doi: 10.1073/pnas.0408836102.
20. Lin W, Watanabe H, Peng S, Francis JM, Kaplan N, Pedamallu CS, Ramachandran A, Agoston A, Bass AJ, Meyerson M. Dynamic epigenetic regulation by menin during pancreatic islet tumor formation. Mol Cancer Res. 2015; 13:689-98. doi: 10.1158/1541-7786.MCR-14-0457.
21. Perren A, Wiesli P, Schmid S, Montani M, Schmitt A, Schmid C, Moch H, Komminoth P. Pancreatic endocrine tumors are a rare manifestation of the neurofibromatosis type 1 phenotype: molecular analysis of a malignant insulinoma in a NF-1 patient. Am J Surg Pathol. 2006; 30:1047-51.
22. Lesma E, Ancona S, Sirchia SM, Orpianesi E, Grande V, Colapietro P, Chiaramonte E, Di Giulio AM, Gorio A. TSC2 epigenetic defect in primary LAM cells. Evidence of an anchorage-independent survival. J Cell Mol Med. 2014; 18:766-79. doi: 10.1111/jcmm.12237.
23. Robinson CM, Ohh M. The multifaceted von Hippel-Lindau tumour suppressor protein. FEBS Lett. 2014; 588:2704-11. doi: 10.1016/j.febslet.2014.02.026.
24. Gordon M, El-Kalla M, Baksh S. RASSF1 Polymorphisms in Cancer. Mol Biol Int. 2012; 2012:365213. doi: 10.1155/2012/365213.
25. Dammann R, Schagdarsurengin U, Liu L, Otto N, Gimm O, Dralle H, Boehm BO, Pfeifer GP, Hoang-Vu C. Frequent RASSF1A promoter hypermethylation and K-ras mutations in pancreatic carcinoma. Oncogene. 2003; 22:3806-12.
26. House MG, Herman JG, Guo MZ, Hooker CM, Schulick RD, Lillemoe KD, Cameron JL, Hruban RH, Maitra A, Yeo CJ. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg. 2003; 238:423-31.
27. Pizzi S, Azzoni C, Bottarelli L, Campanini N, D’Adda T, Pasquali C, Rossi G, Rindi G, Bordi C. RASSF1A promoter methylation and 3p21.3 loss of heterozygosity are features of foregut, but not midgut and hindgut, malignant endocrine tumours. J Pathol. 2005; 206:409-16.
28. Liu L, Broaddus RR, Yao JC, Xie S, White JA, Wu TT, Hamilton SR, Rashid A. Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod Pathol. 2005; 18:1632-40.
29. Arnold CN, Sosnowski A, Schmitt-Gräff A, Arnold R, Blum HE. Analysis of molecular pathways in sporadic neuroendocrine tumors of the gastro-entero-pancreatic system. Int J Cancer. 2007; 120:2157-64.
30. Malpeli G, Amato E, Dandrea M, Fumagalli C, Debattisti V, Boninsegna L, Pelosi G, Falconi M, Scarpa A. Methylation- associated down-regulation of RASSF1A and up-regulation of RASSF1C in pancreatic endocrine tumors. BMC Cancer. 2011; 11:351. doi: 10.1186/1471-2407-11-351.
31. Serrano J, Goebel SU, Peghini PL, Lubensky IA, Gibril F, Jensen RT. Alterations in the p16INK4a/CDKN2A tumor suppressor gene in gastrinomas. J Clin Endocrinol Metab. 2000; 85:4146-56.
32. de Wilde RF, Heaphy CM, Maitra A, Meeker AK, Edil BH, Wolfgang CL, Ellison TA, Schulick RD, Molenaar IQ, Valk GD, Vriens MR, Borel Rinkes IH, Offerhaus GJ, et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol. 2012; 25:1033-9. doi: 10.1038/modpathol.2012.53.
33. Bartsch DK, Kersting M, Wild A, Ramaswamy A, Gerdes B, Schuermann M, Simon B, Rothmund M. Low frequency of p16(INK4a) alterations in insulinomas. Digestion. 2000; 62:171-7.
34. Habbe N, Bert T, Simon B. Identification of methylation- associated gene expression in neuroendocrine pancreatic tumor cells. Pancreatology. 2007; 7: 352-9.
35. Wild A, Ramaswamy A, Langer P, Celik I, Fendrich V, Chaloupka B, Simon B, Bartsch DK. Frequent methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene in pancreatic endocrine tumors. J Clin Endocrinol Metab. 2003; 88:1367-73. doi: 10.1210/jc.2002-021027.
36. Kulke MH, Hornick JL, Frauenhoffer C, Hooshmand S, Ryan DP, Enzinger PC, Meyerhardt JA, Clark JW, Stuart K, Fuchs CS, Redston MS. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide- based therapy in patients with neuroendocrine tumors. Clin Cancer Res. 2009; 15:338-345. doi: 10.1158/1078-0432.CCR-08-1476.
37. Ekeblad S, Sundin A, Janson ET, Welin S, Granberg D, Kindmark H, Dunder K, Kozlovacki G, Orlefors H, Sigurd M, Oberg K, Eriksson B, Skogseid B. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007; 13:2986-2991. doi: 10.1158/1078-0432.CCR-06-2053.
38. Schmitt AM, Pavel M, Rudolph T, Dawson H, Blank A, Komminoth P, Vassella E, Perren A. Prognostic and predictive roles of MGMT protein expression and promoter methylation in sporadic pancreatic neuroendocrine neoplasms. Neuroendocrinology. 2014; 100:35-44. doi: 10.1159/000365514.
39. Walter T, van Brakel B, Vercherat C, Hervieu V, Forestier J, Chayvialle JA, Molin Y, Lombard-Bohas C, Joly MO, Scoazec JY. O6-Methylguanine-DNA methyltransferase status in neuroendocrine tumours: prognostic relevance and association with response to alkylating agents. Br J Cancer. 2015; 112:523-531. doi: 10.1038/bjc.2014.660.
40. Mei M, Deng D, Liu TH, Sang XT, Lu X, Xiang HD, Zhou J, Wu H, Yang Y, Chen J, Lu CM, Chen YJ. Clinical implications of microsatellite instability and MLH1 gene inactivation in sporadic insulinomas. J Clin Endocrinol Metab. 2009; 94:3448-57. doi: 10.1210/jc.2009-0173.
41. Dejeux E, Olaso R, Dousset B, Audebourg A, Gut IG, Terris B, Tost J. Hypermethylation of the IGF2 differentially methylated region 2 is a specific event in insulinomas leading to loss-of-imprinting and overexpression. Endocr Relat Cancer. 2009; 16:939-52. doi: 10.1677/ERC-08-0331.
42. How-Kit A, Dejeux E, Dousset B, Renault V, Baudry M, Terris B, Tost J. DNA methylation profiles distinguish different subtypes of gastroenteropancreatic neuroendocrine tumors. Epigenomics. 2015; 7: 1245-58. doi: 10.2217/epi.15.85.
43. CaoY, GaoZ, LiL, JiangX, ShanA, CaiJ, PengY, Li Y, Jiang X, Huang X, Wang J, Wei Q, Qin G, Zhao J, Jin X, Liu L, Li Y, Wang W, Wang J, Ning G. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat Commun. 2013; 4: 2810. doi: 10.1038/ncomms3810.
44. Kim JT, Li J, Jang ER, Gulhati P, Rychahou PG, Napier DL, Wang C, Weiss HL, Lee EY, Anthony L, Townsend CM Jr, Liu C, Evers BM. Deregulation of Wnt/β-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinogenesis. 2013; 34: 953-61. doi: 10.1093/carcin/bgt018.
45. Stefanoli M, La Rosa S, Sahnane N, Romualdi C, Pastorino R, Marando A, Capella C, Sessa F, Furlan D. Prognostic relevance of aberrant DNA methylation in g1 and g2 pancreatic neuroendocrine tumors. Neuroendocrinology. 2014; 100: 26-34. doi: 10.1159/000365449.
46. Pipinikas CP, Dibra H, Karpathakis A, Feber A, Novelli M, Oukrif D, Fusai G, Valente R, Caplin M, Meyer T, Teschendorff A, Bell C, Morris TJ, et al. Epigenetic dysregulation and poorer prognosis in DAXX-deficient pancreatic neuroendocrine tumours. Endocr Relat Cancer. 2015; 22: L13-8. doi: 10.1530/ERC-15-0108.
47. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011; 333: 425. doi: 10.1126/science.1207313.
48. Marinoni I, Kurrer AS, Vassella E, Dettmer M, Rudolph T, Banz V, Hunger F, Pasquinelli S, Speel EJ, Perren A. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. 2014; 146: 453- 60. doi: 10.1053/j.gastro.2013.
49. Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S, Calin GA, Volinia S, Liu CG, Scarpa A, Croce CM. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006; 24: 4677-84. DOI: 10.1200/JCO.2005.05.5194.
50. Jiang X, Shan A, Su Y, Cheng Y, Gu W, Wang W, Ning G, Cao Y. miR-144/451 Promote Cell Proliferation via Targeting PTEN/AKT Pathway in Insulinomas. Endocrinology. 2015; 156: 2429-39. doi: 10.1210/en.2014-1966.
51. Modali SD, Parekh VI, Kebebew E, Agarwal SK. Epigenetic regulation of the lncRNA MEG3 and its target c-MET in pancreatic neuroendocrine tumors. Mol Endocrinol. 2015; 29: 224-37. doi: 10.1210/me.2014-1304.
52. Zhang HY, Rumilla KM, Jin L, Nakamura N, Stilling GA, Ruebel KH, Hobday TJ, Erlichman C, Erickson LA, Lloyd RV. Association of DNA methylation and epigenetic inactivation of RASSF1A and beta-catenin with metastasis in small bowel carcinoid tumors. Endocrine. 2006; 30: 299- 306. doi: 10.1007/s12020-006-0008-1.
53. Fotouhi O, Adel Fahmideh M, Kjellman M, Sulaiman L, Höög A, Zedenius J, Hashemi J, Larsson C. Global hypomethylation and promoter methylation in small intestinal neuroendocrine tumors: an in vivo and in vitro study. Epigenetics. 2014; 9: 987-97. doi: 10.4161/epi.28936.
54. Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998; 19: 219-20. doi: 10.1038/890.
55. Rahman MM, Qian ZR, Wang EL, Yoshimoto K, Nakasono M, Sultana R, Yoshida T, Hayashi T, Haba R, Ishida M, Okabe H, Sano T. DNA methyltransferases 1, 3a, and 3b overexpression and clinical significance in gastroenteropancreatic neuroendocrine tumors. Hum Pathol. 2010; 41: 1069-78. doi: 10.1016/j.humpath.2010.01.011.
56. Edfeldt K, Ahmad T, Åkerström G, Janson ET, Hellman P, Stålberg P, Björklund P, Westin G. TCEB3C a putative tumor suppressor gene of small intestinal neuroendocrine tumors. Endocr Relat Cancer. 2014; 21: 275-84. doi: 10.1530/ERC-13-0419.
57. Chan AO, Kim SG, Bedeir A, Issa JP, Hamilton SR, Rashid A. CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene. 2003; 22: 924-34. doi: 10.1038/sj.onc.1206123.
58. Choi JD, Lee JS. Interplay between Epigenetics and Genetics in Cancer. Genomics Inform. 2013; 11: 164-73. doi: 10.5808/GI.2013.11.4.164.
59. Choi IS, Estecio MR, Nagano Y, Kim do H, White JA, Yao JC, Issa JP, Rashid A. Hypomethylation of LINE-1 and Alu in well-differentiated neuroendocrine tumors (pancreatic endocrine tumors and carcinoid tumors). Mod Pathol. 2007; 20: 802-10. doi: 10.1038/modpathol.3800825.
60. Stricker I, Tzivras D, Nambiar S, Wulf J, Liffers ST, Vogt M, Verdoodt B, Tannapfel A, Mirmohammadsadegh A. Site- and grade-specific diversity of LINE1 methylation pattern in gastroenteropancreatic neuroendocrine tumours. Anticancer Res. 2012; 32: 3699-706.
61. Verdugo AD, Crona J, Starker L, Stålberg P, Åkerström G, Westin G, Hellman P, Björklund P. Global DNA methylation patterns through an array-based approach in small intestinal neuroendocrine tumors. Endocr Relat Cancer. 2014; 21: L5-7. doi: 10.1530/ERC-13-0481.
62. Warneboldt J, Haller F, Horstmann O, Danner BC, Füzesi L, Doenecke D, Happel N. Histone H1x is highly expressed in human neuroendocrine cells and tumours. BMC Cancer. 2008; 8: 388. doi: 10.1186/1471-2407-8-388.
63. Magerl C, Ellinger J, Braunschweig T, Kremmer E, Koch LK, Höller T, Büttner R, Lüscher B, Gütgemann I. H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1. Hum Pathol. 2010; 41: 181-9. doi: 10.1016/j.humpath.2009.08.007.
64. Ruebel K, Leontovich AA, Stilling GA, Zhang S, Righi A, Jin L, Lloyd RV. MicroRNA expression in ileal carcinoid umors: downregulation of microRNA-133a with tumor progression. Mod Pathol. 2010; 23: 367-75. doi: 10.1038/modpathol.2009.161.
65. Li SC, Essaghir A, Martijn C, Lloyd RV, Demoulin JB, Oberg K, Giandomenico V. Global microRNA profiling of well-differentiated small intestinal neuroendocrine tumors. Mod Pathol. 2013; 26: 685-96. doi: 10.1038/modpathol.2012.216.
66. Døssing KB, Binderup T, Kaczkowski B, Jacobsen A, Rossing M, Winther O, Federspiel B, Knigge U, Kjær A, Friis-Hansen L. Down-Regulation of miR-129-5p and the let-7 Family in Neuroendocrine Tumors and Metastases Leads to Up-Regulation of Their Targets Egr1, G3bp1, Hmga2 and Bach1. Genes (Basel). 2014; 6: 1-21. doi: 10.3390/genes6010001.
67. Pelosi G, Fumagalli C, Trubia M, Sonzogni A, Rekhtman N, Maisonneuve P, Galetta D, Spaggiari L, Veronesi G, Scarpa A, Malpeli G, Viale G. Dual role of RASSF1 as a tumor suppressor and an oncogene in neuroendocrine tumors of the lung. Anticancer Res. 2010; 30: 4269-81.
68. Chaussade L, Eymin B, Brambilla E, Gazzeri S. Expression of p15 and p15.5 products in neuroendocrine lung tumours: relationship with p15(INK4b) methylation status. Oncogene. 2001; 20: 6587-96. doi: 10.1038/sj.onc.1204798.
69. Shilo K, Wu X, Sharma S, Welliver M, Duan W, Villalona- Calero M, Fukuoka J, Sif S, Baiocchi R, Hitchcock CL, Zhao W, Otterson GA. Cellular localization of protein arginine methyltransferase-5 correlates with grade of lung tumors. Diagn Pathol. 2013; 8:201. doi: 10.1186/1746-1596-8-201.
70. Findeis-Hosey JJ, Huang J, Li F, Yang Q, McMahon LA, Xu H. High-grade neuroendocrine carcinomas of the lung highly express enhancer of zeste homolog 2, but carcinoids do not. Hum Pathol. 2011; 42: 867-72. doi: 10.1016/j.humpath.2010.09.019.
71. Li F, Ye B, Hong L, Xu H, Fishbein MC. Epigenetic modifications of histone h4 in lung neuroendocrine tumors. Appl Immunohistochem Mol Morphol. 2011; 19: 389-94. doi: 10.1097/PAI.0b013e3182108e2e.
72. Deng B, Molina J, Aubry MC, Sun Z, Wang L, Eckloff BW, Vasmatzis G, You M, Wieben ED, Jen J, Wigle DA, Yang P. Clinical biomarkers of pulmonary carcinoid tumors in never smokers via profiling miRNA and target mRNA. Cell Biosci. 2014; 4: 35. doi: 10.1186/2045-3701-4-35.
73. Mairinger FD, Walter RF, Theegarten D, Hager T, Vollbrecht C, Christoph DC, Worm K, Ting S, Werner R, Stamatis G, Mairinger T, Baba H, Zarogoulidis K, et al. Gene Expression Analysis of the 26S Proteasome Subunit PSMB4 Reveals Significant Upregulation, Different Expression and Association with Proliferation in Human Pulmonary Neuroendocrine Tumours. J Cancer. 2014; 5: 646-54. doi: 10.7150/jca.9955.
74. Rapa I, Votta A, Felice B, Righi L, Giorcelli J, Scarpa A, Speel EJ, Scagliotti GV, Papotti M, Volante M. Identification of MicroRNAs Differentially Expressed in Lung Carcinoid Subtypes and Progression. Neuroendocrinology. 2015; 101: 246-55. doi: 10.1159/000381454.
75. Lee HW, Lee EH, Ha SY, Lee CH, Chang HK, Chang S, Kwon KY, Hwang IS, Roh MS, Seo JW. Altered expression of microRNA miR-21, miR-155, and let-7a and their roles in pulmonary neuroendocrine tumors. Pathol Int. 2012; 62: 583-91. doi: 10.1111/j.1440-1827.2012.02845.x.
76. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015; 372: 793-5. doi: 10.1056/NEJMp1500523.
77. Caplin ME, Pavel M, Ćwikła JB, Phan AT, Raderer M, Sedláková E, Cadiot G, Wolin EM, Capdevila J, Wall L, Rindi G, Langley A, Martinez S, et al; CLARINET Investigators. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014; 371: 224-33. doi: 10.1056/NEJMoa1316158.
78. Yao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, Tomasek J, Raderer M, Lahner H, Voi M, Pacaud LB, Rouyrre N, Sachs C, et al; RAD001 in Advanced Neuroendocrine Tumours, Fourth Trial (RADIANT-4) Study Group. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016; 387: 968-77. doi: 10.1016/S0140-6736(15)00817-X.
79. Karlsson A, Jönsson M, Lauss M, Brunnström H, Jönsson P, Borg Å, Jönsson G, Ringnér M, Planck M, Staaf J. Genome-wide DNA methylation analysis of lung carcinoma reveals one neuroendocrine and four adenocarcinoma epitypes associated with patient outcome. Clin Cancer Res. 2014; 20: 6127-40. doi: 10.1158/1078-0432.CCR-14-1087.
80. Mairinger FD, Ting S, Werner R, Walter RF, Hager T, Vollbrecht C, Christoph D, Worm K, Mairinger T, Sheu-Grabellus SY, Theegarten D, Schmid KW, Wohlschlaeger J. Different micro-RNA expression profiles distinguish subtypes of neuroendocrine tumors of the lung: results of a profiling study. Mod Pathol. 2014; 27: 1632-40. doi: 10.1038/modpathol.2014.74.
81. Sadanandam A, Wullschleger S, Lyssiotis CA, Grötzinger C, Barbi S, Bersani S, Körner J, Wafy I, Mafficini A, Lawlor RT, Simbolo M, Asara JM, Bläker H, et al. A Cross-Species Analysis in Pancreatic Neuroendocrine Tumors Reveals Molecular Subtypes with Distinctive Clinical, Metastatic, Developmental, and Metabolic Characteristics. Cancer Discov. 2015; 5: 1296-313. doi: 10.1158/2159-8290.CD-15-0068.
82. Cives M, Rizzo F, Simone V, Bisceglia F, Stucci S, Seeber A, Spizzo G, Montrone T, Resta L, Silvestris F. Reviewing the Osteotropism in Neuroendocrine Tumors: The Role of Epithelial-Mesenchymal Transition. Neuroendocrinology. 2015; 103: 321-34. doi: 10.1159/000438902.
83. Guo F, Wang Y, Liu J, Mok SC, Xue F, Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. 2016; 35: 816-26. doi: 10.1038/onc.2015.139.
84. Kidd M, Drozdov I, Modlin I. Blood and tissue neuroendocrine tumor gene cluster analysis correlate, define hallmarks and predict disease status. Endocr Relat Cancer. 2015; 22: 561-75. doi: 10.1530/ERC-15-0092.
85. Lee YS, Kim H, Kim HW, Lee JC, Paik KH, Kang J, Kim J, Yoon YS, Han HS, Sohn I, Cho J, Hwang JH. High Expression of MicroRNA-196a Indicates Poor Prognosis in Resected Pancreatic Neuroendocrine Tumor. Medicine (Baltimore). 2015; 94: e2224. doi: 10.1097/MD.0000000000002224.
86. Shukla S, Meeran SM. Epigenetics of cancer stem cells: Pathways and therapeutics. Biochim Biophys Acta. 2014; 1840: 3494-3502. doi: 10.1016/j.bbagen.2014.09.017.
87. Alexander VM, Roy M, Steffens KA, Kunnimalaiyaan M, Chen H. Azacytidine induces cell cycle arrest and suppression of neuroendocrine markers in carcinoids. Int J Clin Exp Med. 2010; 3: 95-102.
88. Baradari V, Huether A, Höpfner M, Schuppan D, Scherübl H. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocr Relat Cancer. 2006; 13: 1237-50. doi: 10.1677/erc.1.01249.
89. Arvidsson Y, Johanson V, Pfragner R, Wängberg B, Nilsson O. Cytotoxic Effects of Valproic Acid on Neuroendocrine Tumour Cells. Neuroendocrinology. 2015; doi: 10.1159/000441849.
90. Greenblatt DY, Vaccaro AM, Jaskula-Sztul R, Ning L, Haymart M, Kunnimalaiyaan M, Chen H. Valproic acid activates notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist. 2007; 12: 942-51. doi: 10.1634/theoncologist.12-8-942.
91. Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, Chen H. Overexpression of the NOTCH1 intracellular domain inhibits cell proliferation and alters the neuroendocrine phenotype of medullary thyroid cancer cells. J Biol Chem. 2006; 281: 39819-30. doi: 10.1074/jbc.M603578200.
92. Sun L, Qian Q, Sun G, Mackey LV, Fuselier JA, Coy DH, Yu CY. Valproic acid induces NET cell growth arrest and enhances tumor suppression of the receptor-targeted peptide-drug conjugate via activating somatostatin receptor type II. J Drug Target. 2016; 24: 169-77. doi: 10.3109/1061186X.2015.1066794.
93. Adler JT, Hottinger DG, Kunnimalaiyaan M, Chen H. Combination therapy with histone deacetylase inhibitors and lithium chloride: a novel treatment for carcinoid tumors. Ann Surg Oncol. 2009; 16: 481-6. doi: 10.1245/s10434-008-0194-6.
94. Kusunoki M, Yamamura T, Ichii S, Fujita S, Nakai T, Utsunomiya J. The effects of sodium valproate on plasma somatostatin and insulin in humans. J Clin Endocrinol Metab. 1988; 67:1060-3. doi: 10.1210/jcem-67-5-1060.
95. Mohammed TA, Holen KD, Jaskula-Sztul R, Mulkerin D, Lubner SJ, Schelman WR, Eickhoff J, Chen H, Loconte NK. A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist. 2011; 16: 835-43. doi: 10.1634/theoncologist.2011-0031.
96. Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006; 12: 3997-4003. doi: 10.1158/1078-0432.CCR-05-2689.
97. Rajguru S, Lubner SJ, Mulkerin D, Schelman WR, Winterle N, Holen KD, Leverson G, Chen H, LoConte NK. A phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in low-grade neuroendocrine tumors. J Clin Oncol. 2012; 30: abstr e14554 (suppl).
98. Yao JC, Lagunes DR, Kulke MH. Targeted therapies in neuroendocrine tumors (NET): clinical trial challenges and lessons learned. Oncologist. 2013; 18: 525-32. doi: 10.1634/theoncologist.2012-0434.
99. Carter CA, Degesys A, Oronsky B, Scicinski J, Caroen SZ, Oronsky AL, Reid T, Cabrales P, Roswarski J. Flushing Out Carcinoid Syndrome: Beneficial Effect of the Anticancer Epigenetic Agent RRx-001 in a Patient with a Treatment-Refractory Neuroendocrine Tumor. Case Rep Oncol. 2015; 8: 461-5. doi: 10.1159/000441775.
100. Boora GK, Kanwar R, Kulkarni AA, Pleticha J, Ames M, Schroth G, Beutler AS, Banck MS. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet. 2015; 208: 374-81. doi: 10.1016/j.cancergen.2015.04.002.
101. Vandamme T, Peeters M, Dogan F, Pauwels P, Van Assche E, Beyens M, Mortier G, Vandeweyer G, de Herder W, Van Camp G, Hofland LJ, Op de Beeck K. Whole-exome characterization of pancreatic neuroendocrine tumor cell lines BON-1 and QGP-1. J Mol Endocrinol. 2015; 54: 137- 47. doi: 10.1530/JME-14-0304.
102. Angermueller C, Clark SJ, Lee H, Macaulay IC, Teng MJ, Hu TX, Krueger F, Smallwood SA, Ponting CP, Voet T, Kelsey G, Stegle O, Reik W. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016; 13:229-32. doi: 10.1038/nmeth.3728.
103. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, Makarov V, Buhu S, Slamon DJ, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015; 162: 974-86. doi: 10.1016/j.cell.2015.07.011.
104. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, O’Brien C, De Carvalho DD. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell. 2015; 162: 961-73. doi: 10.1016/j.cell.2015.07.056.
105. Weintraub K. Take two: Combining immunotherapy with epigenetic drugs to tackle cancer. Nat Med. 2016; 22: 8-10. doi: 10.1038/nm0116-8.