
INTRODUCTION
The global prevalence of diabetes has risen to be 9% among adults aged over 18 years in 2014 [1]. T2D comprises around 90% of people with diabetes in the world [2]. It is a worldwide concern that more than half a billion people will be affected by this disease by 2030 [3]. An investigation based on sample weighting indicated nearly 113.9 million Chinese adults suffering from diabetes and 493.4 million people with prediabetes [4].
The development of T2D is a complex process implicating synergistic effects of genetic susceptibility and environmental factors [5]. Recent studies have unmasked that the status of gastrointestinal tract microbiota has a marked impact on nutrition-related syndromes such as obesity and T2D [6]. Although genetic factors promote the susceptibility of metabolic disease, the contribution of GI tract microbes as potential partaker in the development of T2D cannot be ignored. Signal molecules produced by microbial metabolism stimulate the pancreatic insulin secretion, improve insulin sensitivity and vary insulin signaling [7], or motivate intestinal gluconeogenesis benefiting energy and glucose homeostasis [8]. A large number of bacterial translocation contribute to insulin resistance by disturbing insulin receptor modification and inhibiting the binding of insulin to its receptor [9]. Recently, the next generation sequencing Illumina Miseq of 16S rRNA gene libraries driven by high-throughput technologies has been used to characterize the diversity of bacterial communities [10]. It is an initially crucial process to assemble short sequences into operational taxonomic units (OTUs) in analyzing metagenomic data [11]. We employed the method above to evaluate whether the properties of different GI sections utilize essential selective pressures on microbiota and play critical roles in the forming of GI microbiota.
Zucker diabetic fatty (ZDF) rats with a missense mutation (fatty, fa) in the leptin receptor gene (LEPR) develop obesity, insulin resistance, and T2D [12-15]. The male ZDF rats present an age-dependent diabetes phenotype, which develop hyperglycemia by 8 weeks of age with serum glucose levels remaining high throughout its life-span [16]. By virtue of these features, the ZDF rat is an attractive experimental model for this study. In this study, we characterized the GI tract microbes of specific pathogen free (SPF) ZDF rats using a high-throughput 16S rRNA sequencing technology.
RESULTS
Body weight and blood glucose levels
All rats gained weight during the experiment. Body weight of ZL rats was significantly lower at baseline and in the end of this study compared to ZDF rats, and less body weight was gained in the ZL group compared to the ZDF group (Figure 1A). The blood glucose levels were no significant difference between ZL group and ZDF group at first. From week 2, the levels of blood glucose were continuously elevated in ZDF rats (Figure 1B).
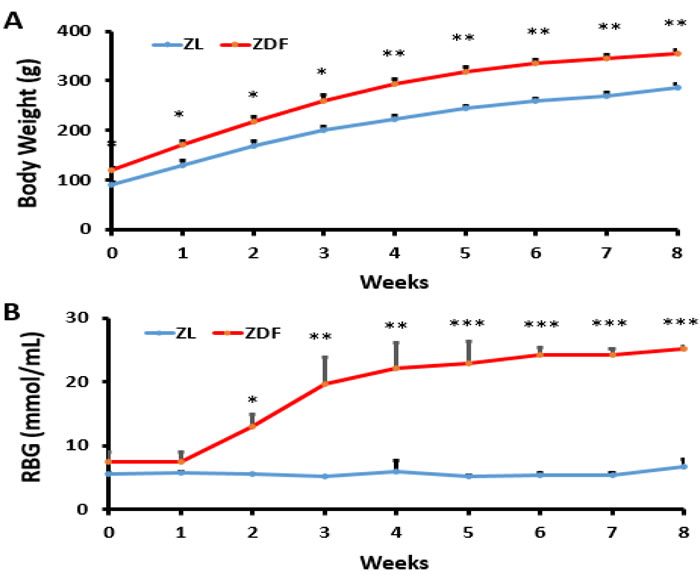
Figure 1: Body weight and blood glucose levels. A. Body weight of ZDF rats (Zucker diabetic fatty rats) was significantly higher than ZL rats (ZDF-lean rats). *p < 0.05, **p < 0.01, ZDF vs. ZL B. Random blood glucose of ZDF rats was significantly higher than ZL rats. *p < 0.05, **p < 0.01, ***p < 0.001, ZDF vs. ZL.
High-throughput 16S rRNA sequencing along the rat GI tract
A total of 840,576 sequence reads with a mean length of 344.2 ± 5.49 bp (mean ± SD) were obtained from all samples (stomach, duodenum, jejunum, ileum, cecum and feces) in ZDF rats and the lean littermates (ZL rats) by Miseq Sequencing analysis. Each sample was covered by an average of 24,016 reads. Except one duodenum sample from a ZDF rat containing a very low number of sequences, which was removed from the diversity analysis, all rests met the requirements for library establishment (Table S1 in File S1). The individual rarefaction curves tended to approach the saturation plateau (Figure 2), suggesting high sampling coverage (~99%) was achieved in all samples.
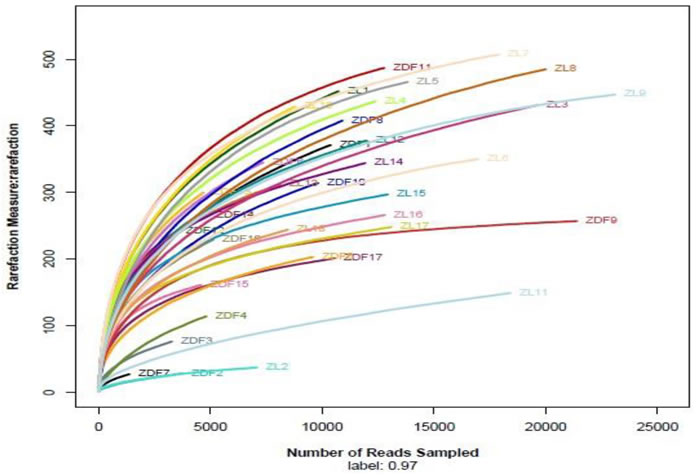
Figure 2: Individual rarefaction curves. ZL rats and ZDF rats, n = 3, respectively. ZL 1-3, stomach; ZL 4-6, duodenum; ZL 7-9, jejunum; ZL 10-12, ileum; ZL 13-15, cecum; ZL 16-18, feces. ZDF 1-3, stomach; ZDF 4-5, duodenum; ZL 6-8, jejunum; ZL 9-11, ileum; ZL 12-14, cecum; ZL 15-17, feces.
OTU network analyses of bacterial communities
OTUs and different GI tract sites of ZDF rats were labeled as nodes in bipartite network. OTUs were linked with the samples, and their sequences would be found in OTU-nodes [17]. As shown in Figure 3, the OTUs network-based analyses displayed that samples from stomach were more closely related to one another from the adjacent part (duodenum) than that from other anatomic sites, which presented that higher similarity of samples between lower digestive tract and feces than stomach and duodenum. Furthermore, ‘‘shared’’ OTUs were found in the same GI tract site collected from different individuals. Different sites shared different common ‘‘core of flora” both in amount and compositions, which might manipulate unique functions from one GI site to other sites. The stomach and jejunum of the three individuals had a small ‘‘core’’ microbiota (16 and 18 OTUs) composed of bacteria belonging to Lactobacillus (Table S2 in File S1), whereas the ileum, cecum and feces of them had a relatively bigger ‘‘core’’ microbiota (81, 79 and 64 OTUs) comprised of bacteria belonging to Clostridia, Bacteroidia, Erysipelotrichia and Bacilli (Table S2 in File S1). Different anatomical sites retain their own unique physicochemical environments, including nutrient supplies, pH, redox potential, intestinal motility, and host secretions [18, 19]. OTU network analyses would provide supports for the hypothesis that the properties of different GI sections utilize essential selective pressures on microbiota and play critical roles in the forming of GI microbiota.
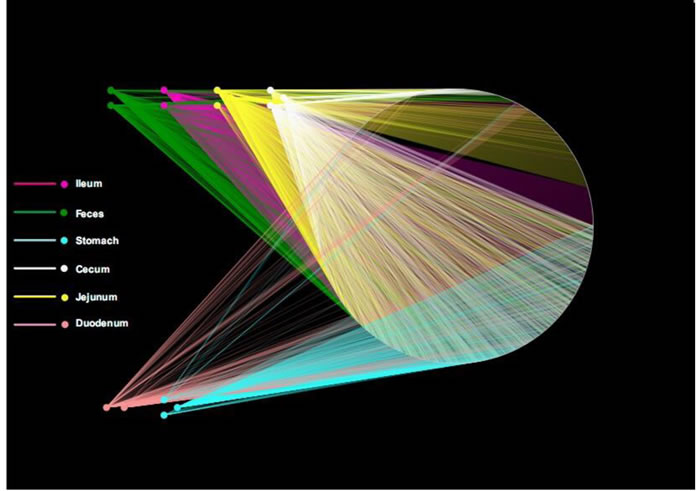
Figure 3: Operational taxonomic unit (OTU) network. OTU network analysis of bacterial communities from ZDF rat GI tract samples for the V3 16S rRNA region.
Diversity of the bacterial community along the ZDF rat GI tract
To characterize gastrointestinal microbes in ZDF rats, bacterial diversity analysis was performed. For alpha diversity analysis, Shannon Index (SI) was estimated to evaluate the diversity of microbes from each sample (Figure 4). In general, stomach samples had the lowest diversity, while samples from cecum and feces had the highest SI values. In addition, the SI values of stomach and small intestine samples showed much higher inter individual variation than those from cecum and fecal samples.
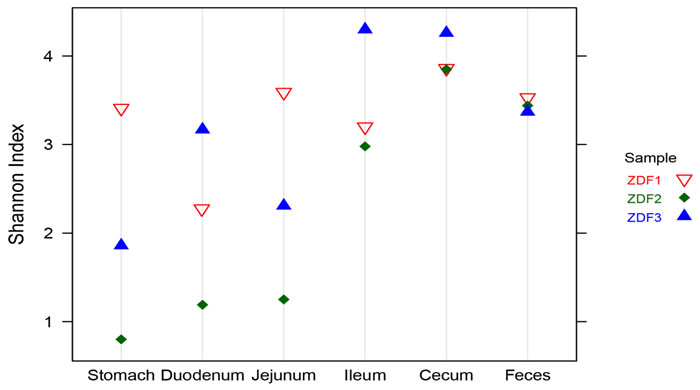
Figure 4: Alpha diversity. Shannon diversity of each GI site from three ZDF rats.
For beta diversity analysis of the ZDF rat GI tract, 20 different bacterial phyla were identified. The communities within the distinct sections of the GI tracts differed largely in their compositions and proportions of the major bacteria. The majority of the sequences belonged to Firmicutes (68.7%) and Bacteroidetes (17.3%), while the rest were Proteobacteria (5.9%), Actinobacteria (4.0%), Tenericutes (3.2%), Verrucomicrobia (0.29%) and unclassified bacteria (0.63%) (Figure 5). Firmicutes was the most abundant phyla in all samples, however, Bacteroidetes was the main bacterial phyla of fecal samples (55.4% ± 0.06). In the 6 GI sites, bacterial structure of cecum and fecal samples were similar, but quite different from other anatomic sites. As shown in Figure 5A, the mucosa of the jejunum contained a comparatively higher proportion of Tenericutes suggesting that Tenericutes may be the new marker for microorganism research in the jejunum of ZDF rat with T2D. At genus level, the facultative bacteria Lactobacillus, belonging to Bacilli (class), Lactobacillaceae (family) was enriched in stomach and small intestine, and decreased from stomach to feces (Figure 5B and 5C). Moreover, genus Lactobacillus representing a heterogeneous group has been well documented of immune modulating properties [20] and might potentially contribute to chronic inflammation in diabetic subjects.
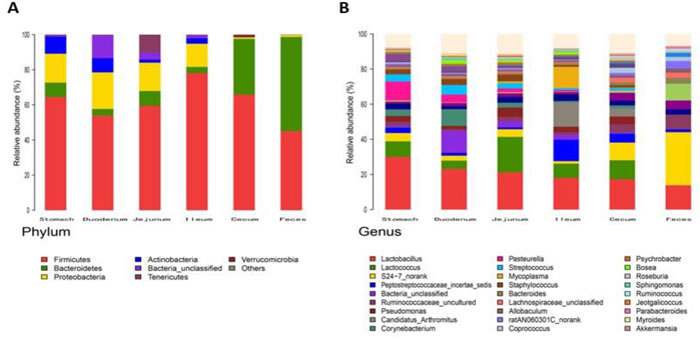
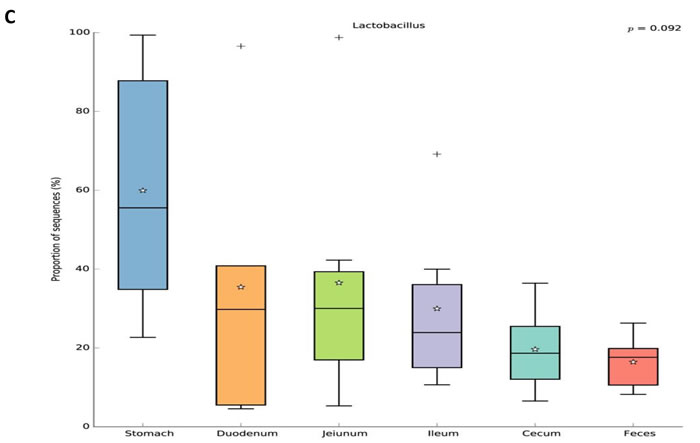
Figure 5: Analysis of Bacterial composition in ZDF rats of each GI site. A. Beta diversity analysis of the ZDF rat GI tract at phyla level. B. Beta diversity analysis of the ZDF rat GI tract at genus level. C. Distribution of Lactobacillus along the ZDF rat GI tract.
Bacterial taxonomic compositions in ZDF rats compared to ZL rats
To explore the effects of missense mutation on the GI tract micro ecology, we compared bacterial communities in different anatomic sites of ZDF rat with its control. At phylum level, in both groups, Firmicutes was dominant in stomach and small intestine, and the dominant bacteria of caecum and feces were Firmicutes and Bacteroidetes. The relative abundance of Firmicutes in 5 major functional compartments of the ZDF rat GI tracts and feces were increased (Figure 6A). Although no statistically significant differences were found in the analyzed abundance of the bacterial groups, Firmicutes were increased in ZDF group compared to ZL group, while Bacteroidetes were decreased in ZDF group compared to ZL group. And the Firmicutes/Bacteroidetes ratio increased in ZDF rats compared to ZL rats, especially in Stomach samples (Table 1). At genus level, Lactobacillus was increased in ZDF group compared to ZL group and decreased from stomach to feces (Figure 6C).
Table 1: Proportion of sequences of Firmicutes and Bacteroidetes along the gastrointestinal tracts in ZDF and ZL rats.
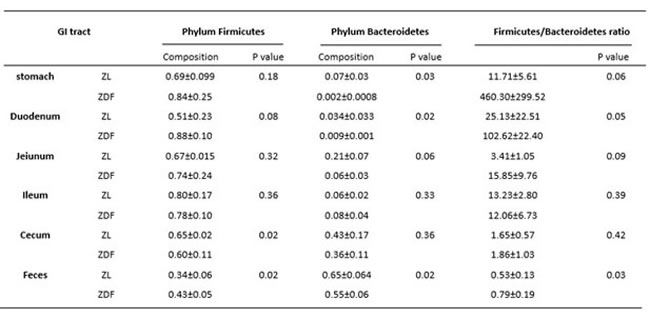
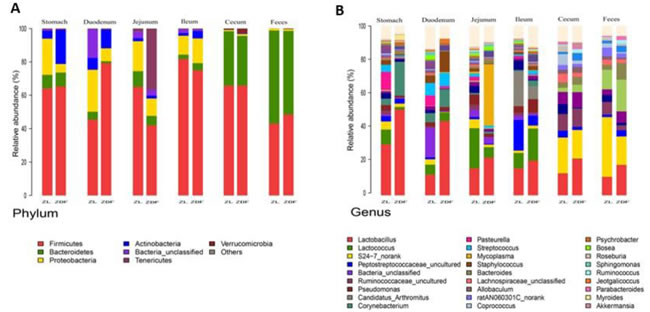
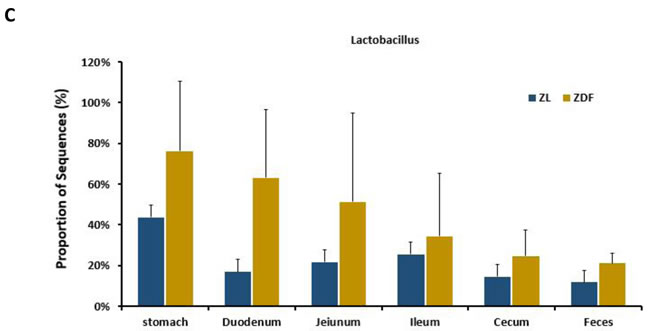
Figure 6: Bacterial taxonomic composition in ZDF rats compared to ZL rats. A. The difference of Bacterial compositions between ZDF and ZL rats at phyla level. B. The difference of Bacterial compositions between ZDF and ZL rats at class level. C. Distribution of Lactobacillus along the ZDF and ZL rat GI tracts.
DISCUSSION
Microbial community in mammalian gastrointestinal (GI) tract plays an important role in overall health and function. Altering human gut microbiota can influence human health, for instance, dietary changes and antibiotic usage may reduce microbiota complexity [21, 22]. This has led to concern that disruption of gut microbiota could improve conditions such as inflammatory bowel disease, obesity, diabetes, metabolic syndrome, cancer, autoimmune disorders [23]. The human gut microbiome maintains a high intra- and inter-subject variability with four dominant phyla of Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria [24]. Up to 90% of the gut microbiota belongs to Firmicutes, Bacteroidetes, and Actinobacteria phyla [25, 26]. Recently, it has been established that the human GM plays crucial roles in type 2 diabetes [27], which demonstrated that a moderate dysbiosis in patients with type 2 diabetes leads to increased risk of a “functional dysbiosis”, rather than a specific microbial species associated directly with T2D pathophysiology.
Numerous metagenomics studies also investigated the gut microbial changes in obesity-the main precursor in the development of T2D in various animal and human studies [28, 29]. Obesity is a major non-communicable global health problem of current era that confers consequential excess risk for T2D [29-31]. Insulin resistance is generally thought to be associated with obesity [32-34]. Typically, T2D is considered to be related to the combination of two metabolic defects, failure of pancreatic beta cells secreting sufficient insulin to compensate for the rising demand and insulin resistance [35, 36]. The latest study reported that GM is a promising modulator of insulin resistance in TLR 2 knockout mice [37]. It is well known that the majority reside microbes in the GI tract modulate host physiology and nutrient intake, and numerous studies focused on the bacterial communities in large intestine and/or feces [38-41].
In order to establish possible associations between the GM changes and T2D, we had conducted the obese diabetic Zucker rat in this investigation, which exhibits hyperglycemia and hyperlipidemia with early onset of insulin resistance. Based on our results, distinctive differences in gut microbiota richness and diversity were observed. In general, stomach samples have the lowest diversity, while samples from cecum and feces have the highest SI values. In addition, the SI values of stomach and small intestine samples showed much higher inter individual variation than those from cecum and fecal samples. Bacterial diversity along the ZDF GI tract was thought to increase from stomach to feces due to stomach and upper small intestine being too harsh (low pH) that makes microorganisms hard to grow and to maintain greater diversity. At phylum level, in both ZDF and ZL groups, Firmicutes was dominant in stomach and small intestine, and the dominant bacteria of caecum and feces were Firmicutes and Bacteroidetes. The relative abundance of Firmicutes in 5 major functional compartments of the ZDF rat GI tract and feces were increased (Figure 6A), in fact, which was also detected in human and mice GI tract [42-45]. Due to limited experimental subjects, the differences of some data are not significant. Although no statistically significant differences were found in the analyzed abundance of the bacterial groups, increased Firmicutes and decreased Bacteroidetes were examined in ZDF group compared to ZL group. And the rising Firmicutes/Bacteroidetes ratio was validated in ZDF rats compared to ZL rats, especially in stomach samples (Table 1). At genus level, Lactobacillus was increased in ZDF group compared to ZL group and decreased from stomach to feces (Figure 6C). Our results support the notion that the higher Firmicutes/Bacteroidetes ratio and the increased levels of Lactobacillus are essential to the prevalence of obesity [46-49]. Similar report claimed that the gut microbiota in T2D patients is characterized by greater numbers of Lactobacillus and reduced Bifidobacterium species [50]. Our work is consistent with above research findings. As numerous reports focus on fecal samples in animal and human studies of T2D, we for the first time systematically characterizes bacterial communities along ZDF rat GI tract to indicate bacterial communities associated to T2D.
In summary, the present study revealed the characterization of gastrointestinal microbes (GM) in ZDF rat GI tract. Although experimental and clinical studies have shown that targeting gut microbiota might be an effective strategy to prevent and manage diabetes [51-53], the concept of using the microbiota as a biomarker of impending or fully manifest T2D within or outside of the GI tract and for monitoring responses to therapeutic interventions needs to be explored.
MATERIALS AND METHODS
Animals
All animal work was performed according to the guidelines of the Institutional Animal Care and local veterinary office and ethics committee of Dalian Medical University (Permit Number: SYXK (Liao) 2008-0002). 5-week-old male ZDF (fa/fa) rats (n = 3) and their age-matched normal lean littermate controls (n = 3) were purchased from Vital River Laboratories (VRL) (Beijing, China) and housed in the specific pathogen-free (SPF) animal experiment center at Dalian Medical University. The rats were fed with high fat food and water (autoclaved before use) ad libitum and housed at 24 °C ± 2 °C with 65% ± 5% humidity on a 12 h light/dark cycle. Adaptive feeding lasted one week.
Random blood glucose test
Random blood glucose (RBG) was measured weekly to examine the development of diabetes in the ZDF rats. Glucose levels in tail blood samples were determined using a glucometer (Roche, Mannheim, Germany) from week 0 to week 8.
Sample preparation
Fresh feces were collected in sterile tube and stored at -80OC immediately. The animals were anesthetized with ether and decapitated before the stomach, duodenum, jejunum, ileum, and cecum were sampled and weighed. Then all samples (36 in total) were snap-frozen in liquid nitrogen and stored at -80°C. The mean lengths of small intestine (including duodenum, jejunum and ileum) and cecum were 6 and 8 cm respectively.
DNA extraction and PCR amplification
Genomic DNA was extracted from stomach, duodenum, jejunum, ileum, cecum and feces samples using the E.Z.N.A.® Stool DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) according to manufacturer’s protocols. The V3 regions of the bacteria 16S ribosomal RNA genes were amplified genomic DNA by PCR (95 °C for 2 min, followed by 27 cycles at 95 °C for 30 s, 55°C for 30 s, and 72°C for 45 s and a final extension at 72 °C for 10 min, 10°C until halted by user) using primers 27F 5’-barcode- AGAGTTTGATCCTGGCTCAG-3’ and 533R 5’-AGAGTTTGATCCTGGCTCAG -3’. 8-bp barcode sequence unique to each samples was attached into forward primer for multiplexing. PCR reactions were performed in triplicate 20 μL reaction mixtures containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.4 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 15 ng of template DNA.
Illumina MiSeq sequencing
The PCR products were separated by 2% agarose gel electrophoresis and bands of the desired size (approximately 250 bp) were purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, U.S.) according to the manufacturer’s instructions. Prior to sequencing, the DNA concentration of each PCR product was determined using QuantiFluor™ -ST (Promega, U.S.). The equimolar purified products were pooled and paired-end sequenced (2 × 250) on an Illumina MiSeq platform according to the standard protocols at Majorbio Bio-Pharm Technology (Shanghai, China) and Lingen Biotechnology Co., Ltd. (Shanghai, China).
Process of sequencing data
Three criteria were followed for demultiplexing and quality-filtering the raw fastq files by QIIME (version 1.17) [54, 55]. Operational taxonomic unit (OTU)-based method was performed to analyze where sequences were split into bins on the basis of taxonomy and clustered to each bin with the cutoff point of 0.05. UPARSE was used to cluster OTUs with 97% similarity cutoff and UCHIME was applied to identify and remove chimeric sequences. The phylogenetic affiliation analysis of each 16S rRNA gene sequence was introduced by RDP Classifier against the silva (SSU115)16S rRNA database with confidence threshold of 70% [56, 57].
Diversity analysis
In alpha diversity analysis, Chao 1 and ACE [58] were calculated to estimate community richness. The Simpson index and Shannon index-based measurements were used to evaluate community evenness. Estimators of community richness, evenness and diversity were calculated based on OTUs (97% similarity).
Statistical analysis
Data are expressed as mean ± SD. Statistical analysis of difference among groups was performed by two-trail Student’s test using SPSS Statistics 18.0 (IBM, New York, USA). Variables with non-Gaussian distribution were ASIN-square-root-transformed for comparisons. P < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This study was supported by grants from the National Nature Science Foundation of China (No. 81230084), by the Specialized Research Fund for the Doctoral Program of Higher Education of China (20132105130001).
Conflicts of interests
There is no conflict of interest.
REFERENCES
1. Mendis S, Davis S and Norrving B. Organizational update: the world health organization global status report on noncommunicable diseases 2014; one more landmark step in the combat against stroke and vascular disease. Stroke. 2015; 46:e121-122.
2. Alberti KG and Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabetic medicine. 1998; 15:539-553.
3. Klein MS and Shearer J. Metabolomics and Type 2 Diabetes: Translating Basic Research into Clinical Application. Journal of diabetes research. 2016; 2016:3898502.
4. Xu Y, Wang L, He J, Bi Y, Li M, Wang T, Wang L, Jiang Y, Dai M, Lu J, Xu M, Li Y, Hu N, Li J, Mi S, Chen CS, et al. Prevalence and control of diabetes in Chinese adults. Jama. 2013; 310:948-959.
5. Riserus U, Willett WC and Hu FB. Dietary fats and prevention of type 2 diabetes. Progress in lipid research. 2009; 48:44-51.
6. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF and Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:15718-15723.
7. Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR and Delzenne NM. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007; 50:2374-2383.
8. De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, Backhed F and Mithieux G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell. 2014; 156:84-96.
9. Musso G, Gambino R and Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded? Diabetes care. 2010; 33:2277-2284.
10. Mancuso FP, D’Hondt S, Willems A, Airoldi L and De Clerck O. Diversity and Temporal Dynamics of the Epiphytic Bacterial Communities Associated with the Canopy-Forming Seaweed Cystoseira compressa (Esper) Gerloff and Nizamuddin. Frontiers in microbiology. 2016; 7:476.
11. Wei ZG and Zhang SW. MtHc: a motif-based hierarchical method for clustering massive 16S rRNA sequences into OTUs. Molecular bioSystems. 2015; 11:1907-1913.
12. Yokoi N, Hoshino M, Hidaka S, Yoshida E, Beppu M, Hoshikawa R, Sudo K, Kawada A, Takagi S and Seino S. A Novel Rat Model of Type 2 Diabetes: The Zucker Fatty Diabetes Mellitus ZFDM Rat. Journal of diabetes research. 2013; 2013:103731.
13. da Silva BA, Bjorbaek C, Uotani S and Flier JS. Functional properties of leptin receptor isoforms containing the gln-->pro extracellular domain mutation of the fatty rat. Endocrinology. 1998; 139:3681-3690.
14. Yamashita T, Murakami T, Iida M, Kuwajima M and Shima K. Leptin receptor of Zucker fatty rat performs reduced signal transduction. Diabetes. 1997; 46:1077-1080.
15. Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CJ and Hess JF. Leptin receptor missense mutation in the fatty Zucker rat. Nature genetics. 1996; 13:18-19.
16. de Lemos ET, Reis F, Baptista S, Pinto R, Sepodes B, Vala H, Rocha-Pereira P, Silva AS and Teixeira F. Exercise training is associated with improved levels of C-reactive protein and adiponectin in ZDF (type 2) diabetic rats. Medical science monitor. 2007; 13:Br168-174.
17. DeSantis TZ, Dubosarskiy I, Murray SR and Andersen GL. Comprehensive aligned sequence construction for automated design of effective probes (CASCADE-P) using 16S rDNA. Bioinformatics (Oxford, England). 2003; 19:1461-1468.
18. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R and Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Science translational medicine. 2009; 1:6ra14.
19. Hopkins MJ, Sharp R and Macfarlane GT. Variation in human intestinal microbiota with age. Digestive and liver disease. 2002; 34 Suppl 2:S12-18.
20. Zeuthen LH, Christensen HR and Frokiaer H. Lactic acid bacteria inducing a weak interleukin-12 and tumor necrosis factor alpha response in human dendritic cells inhibit strongly stimulating lactic acid bacteria but act synergistically with gram-negative bacteria. Clinical and vaccine immunology. 2006; 13:365-375.
21. Maurice CF, Haiser HJ and Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013; 152:39-50.
22. Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, Henrissat B, Knight R and Gordon JI. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science (New York, NY). 2011; 332:970-974.
23. Cho I and Blaser MJ. The human microbiome: at the interface of health and disease. Nature reviews Genetics. 2012; 13:260-270.
24. Haange SB and Jehmlich N. Proteomic interrogation of the gut microbiota: potential clinical impact. Expert review of proteomics. 2016; 13:535-7. doi: 10.1080/14789450.2016.1190652.
25. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE and Relman DA. Diversity of the human intestinal microbial flora. Science (New York, NY). 2005; 308:1635-1638.
26. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010; 464:59-65.
27. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012; 490:55-60.
28. Mallappa RH, Rokana N, Duary RK, Panwar H, Batish VK and Grover S. Management of metabolic syndrome through probiotic and prebiotic interventions. Indian journal of endocrinology and metabolism. 2012; 16:20-27.
29. Connaughton RM, McMorrow AM, McGillicuddy FC, Lithander FE and Roche HM. Impact of anti-inflammatory nutrients on obesity-associated metabolic-inflammation from childhood through to adulthood. The Proceedings of the Nutrition Society. 2016; 75:115-124.
30. Zlobine I, Gopal K and Ussher JR. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochimica et biophysica acta. 2016.
31. Leong A, Porneala B, Dupuis J, Florez JC and Meigs JB. Type 2 Diabetes Genetic Predisposition, Obesity, and All-Cause Mortality Risk in the U.S.: A Multiethnic Analysis. Diabetes care. 2016; 39:539-546.
32. Ham M, Choe SS, Shin KC, Choi G, Kim JW, Noh JR, Kim YH, Ryu JW, Yoon KH, Lee CH and Kim JB. Glucose-6-phosphate dehydrogenase deficiency improves insulin resistance with reduced adipose tissue inflammation in obesity. Diabetes. 2016.
33. Martinerie C, Garcia M, Huong Do TT, Antoine B, Moldes M, Dorothee G, Kazazian C, Auclair M, Buyse M, Ledent T, Marchal PO, Fesatidou M, Beisseiche A, Koseki H, Hiraoka S, Chadjichristos CE, et al. NOV/CCN3: A new adipocytokine involved in obesity-associated insulin resistance. Diabetes. 2016.
34. Dehkordi EH, Sedehi M, Shahraki ZG and Najafi R. Effect of folic acid on homocysteine and insulin resistance of overweight and obese children and adolescents. Advanced biomedical research. 2016; 5:88.
35. Samuel VT and Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. The Journal of clinical investigation. 2016; 126:12-22.
36. Savu O, Elian V, Steriade O, Teodoru I, Mihut S, Tacu C, Covic A and Serafinceanu C. The impact of basal insulin analogues on glucose variability in patients with type 2 diabetes undergoing renal replacement therapy for end-stage renal disease. International urology and nephrology. 2016; 48:265-70. doi: 10.1007/s11255-015-1175-x.
37. Caricilli AM, Picardi PK, de Abreu LL, Ueno M, Prada PO, Ropelle ER, Hirabara SM, Castoldi A, Vieira P, Camara NO, Curi R, Carvalheira JB and Saad MJ. Retraction: Gut Microbiota Is a Key Modulator of Insulin Resistance in TLR 2 Knockout Mice. PLoS biology. 2016; 14:e1002479.
38. Fugmann M, Breier M, Rottenkolber M, Banning F, Ferrari U, Sacco V, Grallert H, Parhofer KG, Seissler J, Clavel T and Lechner A. The stool microbiota of insulin resistant women with recent gestational diabetes, a high risk group for type 2 diabetes. Scientific reports. 2015; 5:13212.
39. Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Krogh Pedersen H, Arumugam M, Kristiansen K, Voigt AY, Vestergaard H, Hercog R, Igor Costea P, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015; 528:262-266.
40. Bakker GJ, Zhao J, Herrema H and Nieuwdorp M. Gut Microbiota and Energy Expenditure in Health and Obesity. Journal of clinical gastroenterology. 2015; 49 Suppl 1:S13-19.
41. Zhang X, Zhao Y, Xu J, Xue Z, Zhang M, Pang X, Zhang X and Zhao L. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Scientific reports. 2015; 5:14405.
42. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R and Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009; 457:480-484.
43. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD and Gordon JI. Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:11070-11075.
44. Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE and Krajmalnik-Brown R. Human gut microbiota in obesity and after gastric bypass. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:2365-2370.
45. Backhed F, Manchester JK, Semenkovich CF and Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:979-984.
46. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER and Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006; 444:1027-1031.
47. Ley RE, Turnbaugh PJ, Klein S and Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006; 444:1022-1023.
48. Million M, Maraninchi M, Henry M, Armougom F, Richet H, Carrieri P, Valero R, Raccah D, Vialettes B and Raoult D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. International journal of obesity (2005). 2012; 36:817-825.
49. Million M, Angelakis E, Paul M, Armougom F, Leibovici L and Raoult D. Comparative meta-analysis of the effect of Lactobacillus species on weight gain in humans and animals. Microbial pathogenesis. 2012; 53:100-108.
50. Le KA, Li Y, Xu X, Yang W, Liu T, Zhao X, Tang YG, Cai D, Go VL, Pandol S and Hui H. Alterations in fecal Lactobacillus and Bifidobacterium species in type 2 diabetic patients in Southern China population. Frontiers in physiology. 2012; 3:496.
51. Stenman LK, Waget A, Garret C, Klopp P, Burcelin R and Lahtinen S. Potential probiotic Bifidobacterium animalis ssp. lactis 420 prevents weight gain and glucose intolerance in diet-induced obese mice. Beneficial microbes. 2014; 5:437-445.
52. Ogawa A, Kadooka Y, Kato K, Shirouchi B and Sato M. Lactobacillus gasseri SBT2055 reduces postprandial and fasting serum non-esterified fatty acid levels in Japanese hypertriacylglycerolemic subjects. Lipids in health and disease. 2014; 13:36.
53. Candela M, Biagi E, Soverini M, Consolandi C, Quercia S, Severgnini M, Peano C, Turroni S, Rampelli S, Pozzilli P, Pianesi M, Fallucca F and Brigidi P. Modulation of gut microbiota dysbioses in type 2 diabetic patients by macrobiotic Ma-Pi 2 diet. The British journal of nutrition. 2016:1-14.
54. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010; 7:335-336.
55. Sun Q, Li A, Li M and Hou B. Effect of pH on biodiesel production and the microbial structure of glucose-fed activated sludge. International Biodeterioration & Biodegradation. 2015; 104:224-230.
56. Schloss PD and Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Applied and environmental microbiology. 2011; 77:3219-3226.
57. Westcott SL and Schloss PD. De novo clustering methods outperform reference-based methods for assigning 16S rRNA gene sequences to operational taxonomic units. PeerJ. 2015; 3:e1487.
58. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ and Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology. 2009; 75:7537-7541.