
INTRODUCTION
Advanced lung cancer is a lethal disease and very large numbers of patients die annually worldwide [1, 2]. In addition to conventional lung cancer treatments including surgery, radiation and chemotherapy, molecular targeting drugs have recently been developed for treatment of lung cancer and have shown some prognostic advantages [3, 4]; however, the merits for patients remain limited [5, 6]. Elucidation of the biological properties of lung cancer cells is essential to find a cure for lung cancer.
Cancer stem-like cells (CSCs)/cancer-initiating cells (CICs) are thought to be major causes of cancer recurrence, distant metastasis and treatment resistance [7]. The idea of CSCs/CICs has a long and winding story [8], but the first evidence for the existence of CSCs/CICs was obtained for leukemia stem cells [9, 10], and the concept of ‘cancer stem cell’ became important in the cancer research field. CSCs/CICs in a solid tumor were first isolated from breast carcinoma with the combination of CD44 and CD24 expression [11]. CSCs/CICs are thought to be located at the top of a hierarchical differentiation model and maintain themselves by self-renewal. CSCs/CICs produce differentiated non-CSCs/CICs at the same time to form a heterogenic cancer population [12]. According to a cancer stem cell hypothesis, only a fraction of cancer cells have strong clonogenic ability and give rise to a predictable hierarchical model of tumor growth [13]. On the other hand, some groups have recently demonstrated that non-CSCs/CICs can acquire stem-like properties in breast cancer, and those groups obtained proof that cell plasticity is maintained without genetic manipulation and that any subpopulation of cells will return to equilibrium phenotypic proportions over time [14–16]. Plasticity of non-CSC/CIC in colon cancers has also been described [17]. Lung cancer is a highly metastatic disease and treatments often fail due to recurrence, suggesting that CSCs/CICs might play a role in clinical causes. However, plasticity of lung cancer cells has not been determined yet.
Previously, we showed that lung CSCs/CICs can be isolated as side population (SP) cells and that the transcription factor SOX2 is expressed in lung CSCs/CICs [18]. SOX2 has an essential role in the maintenance of lung CSCs/CICs. In this study, we investigated the plasticity of lung CSCs/CICs by using SOX2 as a lung CSCs/CICs marker and we found a novel mechanism of dedifferentiation of lung cancer cells.
RESULTS
Differentiated lung cancer cells dedifferentiate into cancer stem-like cells
In a previous study, we succeeded in isolating lung CSCs/CICs from the lung adenocarcinoma cell line LHK2 as side population (SP) cells [18]. In the present study, we analyzed the self-renewal and differentiation abilities of LHK2 SP cells and main population (MP) cells. SP cells showed higher tumor-initiating ability as described previously [18], and SP cell showed higher expressions of stem cell-related genes including SOX2, ALDH1A1, KLF4 and NANOG (Supplementary Figure S1), indicating that SP cells are enriched with CSCs/CICs. Isolated SP cells and MP cells derived from LHK2 cells were cultured in vitro for 2 weeks, and then the cultured SP cells and MP cells were re-analyzed (Figure 1A). Cultured SP cells included a large percentage of SP cells (29.7%). Furthermore, some of the cultured SP cells had differentiated into MP cells, indicating that SP cells have both self-renew ability and differentiation ability. Interestingly, the proportion of SP cells in cultured MP cells was only 0.06% (Figure 1A). For detailed analysis, we investigated the differentiation status at the single cell level. Single cells were sorted from both SP cells and MP cells and cultured for more than one month until clone cells show stable growth. Several clones were established from both SP cells and MP cells, and clone cells were re-analyzed by an SP assay. Clones derived from SP cells were positive for SP cells (SP rates were 5.04% for SP clone B, 2.19% for SP clone D and 5.96% for SP clone H.) (Figure 1B). Interestingly, clones derived from MP cells were also positive for SP cells (SP rates were 9.67% for MP clone D, 5.13% for MP clone H and 1.03% for MP clone I.). Furthermore, we re-established MP clones and SP clones from one MP clone cells (MP-D). Both SP clones and MP clones derived from MP-D clone cells were positive for SP cells (Figure 1B). To confirm the phenomenon, we performed similar single cell sorting analysis using lung squamous cell carcinoma cell line, Sq-1. Both SP clone cells and MP clone cells showed positive for SP cells (Supplementary Figure S2). These results indicated that lung differentiated MP cells can dedifferentiate into SP cells.
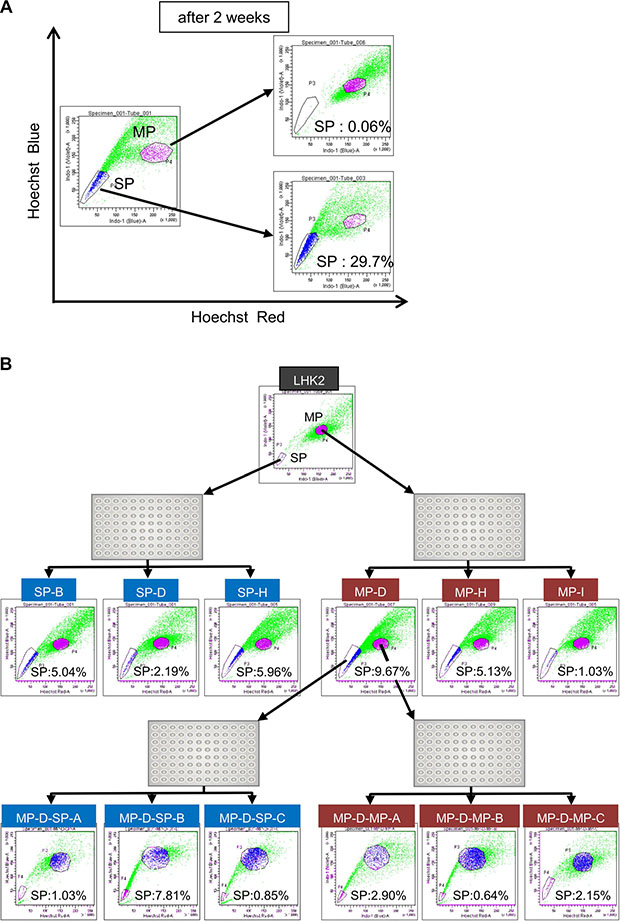
Figure 1: Differentiated non-CSCs/CICs dedifferentiate into CSCs/CICs. (A) SP assay of LHK2 cells. The percentages represent ratios of SP cells and MP cells. Sorted SP cells and MP cells were cultured in DMEM supplemented with 10% FBS for 2 weeks and analyzed by the SP assay again. (B) SP assay of LHK2 SP clone cells and MP clone cells, and second generation of SP clone cells and MP clone cells derived from MP-D clone cells. The percentage represents ratio of SP cells.
SOX2 expression and stemness were regulated by class I HDAC
Previously, we showed that SOX2 was expressed in LHK2 SP cells at a higher level than that in LHK2 MP cells and that SOX2 was involved in the maintenance of lung CSCs/CICs [18]. We thus investigated SOX2 expression levels in LHK2 SP clone cells and MP clone cells by qRT-PCR. SP clone cells showed a significantly higher expression level of SOX2 than that in MP clone cells, and MP clone cells showed low SOX2 expression levels as in MP cells (Figure 2A). MP cells and SP cells derived from MP-D cells were also analyzed, and SP cells derived from MP-D cells showed a higher SOX2 expression level than that in MP cells derived from MP-D cells, but the difference was not statistically significant (p = 0.055) (Figure 2B). These results indicate that a relatively high expression level of SOX2 in the population might be important for production of an SP subpopulation.
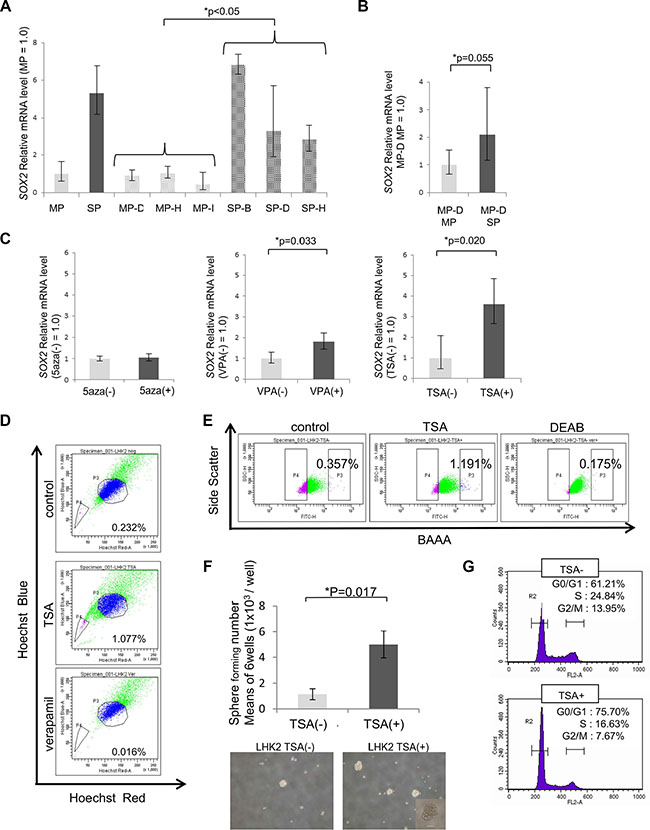
Figure 2: SOX2 expression and stemness are regulated by class I HDAC. (A) Quantitative real-time RT-PCR analysis of SOX2 mRNA expression in LHK2 MP and SP cells, MP clone and SP clone cells, and MP and SP cells derived from MP-D clone cells. MP cells were used for the control, which was set as 1.0. Data are expressed as means ± s.d. of relative values compared with MP cells. Asterisks indicate significant differences. *P < 0.05. Steel-Dwass test. (B) Quantitative real-time PCR analysis of SOX2 mRNA expression in LHK2 MP and SP cells derived from MP-D clone cells. MP cells derived from MP-D cells were used for the control, which was set as 1.0. Data are expressed as means ± s.d. of relative values compared with MP cells. Asterisks indicate significant differences. *P values. Student’s t-test. (C) Quantitative real-time PCR analysis of SOX2 mRNA expression in LHK2 cells treated with 5aza, VPA and TSA. Nontreated cells were used for the control, which was set as 1.0. Data are expressed as means ± s.d. of relative values compared with nontreated cells. Asterisks indicated significant differences. *P values. Paired t-test. (D) SP assay of LHK2 cells treated with TSA. The percentage represents the ratio of SP cells. (E) ALDEFLUOR assay of LHK2 cells treated with TSA. The percentage represents the ratio of ALDHhigh cells. (F) Comparison of the numbers sphere-forming cells not treated with TSA (TSA(–)) and TSA-treated cells (TSA(+)) in LHK2 cells. Asterisks indicated significant differences. *P values. Paired t-test. Images of tumor spheres seeded with TSA(–) and TSA(+) in LHK2 cells. Scale bar, 100 μm. (G) Cell cycle analysis of cells not treated with TSA (TSA–) and TSA-treated cells (TSA+) in LHK2 cells.
Since all of the MP clones of LHK2 showed ability for dedifferentiation into SP cells in just one month of culture, we hypothesized that regulation of dedifferentiation might be controlled by epigenetic regulation, not by genetic events. To investigate the possible epigenetic regulation, we treated LHK2 cells with the DNA methyltransferase inhibitor 5′-aza-2′- deoxycytidine (5aza) and the histone deacetylase (HDAC) inhibitors valproic acid (VPA) and Trichostatin A (TSA) and examined the expression of SOX2 by qRT-PCR. Treatment with 5aza did not change SOX2 expression (Figure 2C). On the other hand, treatments with the HDAC inhibitors VPA and TSA resulted in significant enhancement of SOX2 expression (Figure 2C). Since VPA is a class I HDAC inhibitor and TSA is a class I and class II HDAC inhibitor, SOX2 expression might be controlled by class I HDAC.
To determine whether CSCs/CICs can be induced by an HDAC inhibitor, LHK2 cells were treated with TSA and examined by SP analysis and the ALDEFLUOR assay [22]. The ratio of SP cells was increased by TSA treatment (Figure 2D). Furthermore, aldehyde dehydrogenase 1 (ALDH1) high populations were increased by TSA treatment (Figure 2E). These observations indicate that CSCs/CICs might be induced by TSA treatment. To generalize these phenomena, other lung cancer cell lines of different histological subtypes were analyzed. Lung adenocarcinoma cell line A549, squamous cell carcinoma cell line Sq-1, large cell carcinoma cell line Lu99 and small cell carcinoma cell line Lc817 were treated with TSA, and SOX2 expression and SP cells were investigated. A549 and Sq-1 cells showed significant enhancement of SOX2 expression, and other cell lines also showed a tendency for SOX2 expression enhancement (Supplementary Figure S3A). Furthermore, the ratios of SP cells were increased in all four cell lines by TSA treatment (Supplementary Figure S3B).
Since CSCs/CICs have sphere-forming ability in a floating culture condition [23], we performed a sphere forming assay using LHK2 and Sq-1 cells treated with TSA. Both LHK2 cells and Sq-1 cells showed stronger sphere formation ability in a TSA (+) condition than in a control condition (Figure 2F and Supplementary Figure S3C). Since CSCs/CICs show the quiescent stage in the cell cycle [8], we performed cell cycle analysis using LHK2 cells treated with TSA. TSA-treated LHK2 cells showed a tendency for quiescent stage maintenance (Figure 2G).
The transcription factor HOXA5 induces SOX2 expression in the presence of a class I HDAC inhibitor
SOX2 expression was shown to be regulated by class I HDAC. To further investigate the gene expression mechanisms of SOX2, we searched for putative transcription factor-binding sites in the SOX2 gene promoter region using SABiosciences’ Text Mining Application and UCSC Genome Browser, and we found several candidate transcription factors (Supplementary Figure S4A). We investigated the expression of candidate transcription factors (p300, SOX9, SOX5, POU3F2, FOXL1, HOXA5, zic2 and Nanog) in LHK2 SP cells and MP cells. SOX5, POU3F2, HOXA5 and Nanog showed preferential expression in SP cells (Supplementary Figure S4B). Since the expression of SOX2 was upregulated by class I HDAC inhibitors, regulation of the expression of candidate transcription factors by class I HDAC inhibitors was investigated. SOX5, POU3F2, HOXA5 and zic2 showed enhancement of expression in LHK2 cells treated with class I HDAC inhibitors (Supplementary Figure S4C). To generalize the gene expression in lung cancer cells, A549, Sq-1, Lu99 and Lc817 cells were treated with TSA, and the expression of transcription factors was investigated. Only HOXA5 showed general expression (Supplementary Figure S4D and S4E). Protein expression of HOXA5 was detected in TSA-treated lung cancer cells (Figure 3A and Supplementary Figure S4F). These results indicate that HOXA5 is a possible transcription factor for SOX2 expression, and we thus further analyzed HOXA5. HOXA5 has a single DNA-binding domain, and this gene was barely expressed in some normal adult tissues, lung cancer cell lines and primary lung cancer cells generally (Supplementary Figure S4G).
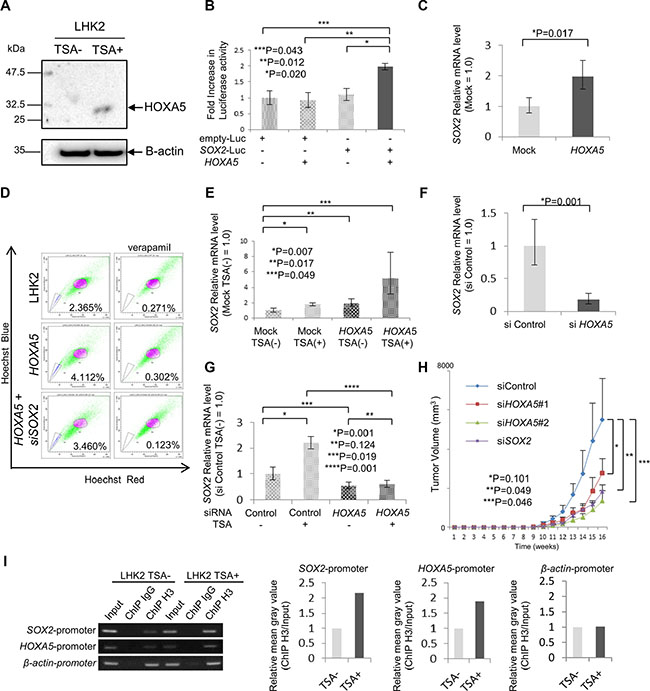
Figure 3: HOXA5 has a role in SOX2 expression and tumorigenicity. (A) Expression of HOXA5 protein in LHK2 cells not treated with TSA (TSA–) and TSA-treated cells (TSA+) confirmed by Western blotting with an anti-HOXA5 antibody. (B) Luciferase assays for extracts of LHK2 cells transfected with HOXA5 expression plasmid plus SOX2 promoter–Luc constructs. The fold increase in Luciferase activity compared with cells transfected with an empty plasmid or empty-Luc plasmid was determined. Data are expressed as means ± s.d. Asterisks indicated significant differences. *P, **P values. Student’s t-test. ***P values. Welch’s t-test. (C) Quantitative real-time RT-PCR analysis of SOX2 mRNA expression in LHK2 cells transfected with HOXA5. Asterisks indicated significant differences. *P values. Paired t-test. (D) SP assay of LHK2 cells transfected with HOXA5 and siSOX2. The percentage represents the ratio of SP cells. (E) Quantitative real-time PCR analysis of SOX2 mRNA expression in empty vector-transfected TSA(–) cells, empty vector-transfected TSA(+) cells, HOXA5-transfected TSA(–) cells and HOXA5-transfected TSA(+) cells. Asterisks indicated significant differences. *P, **P, ***P values. Paired t-test. (F) Quantitative real-time PCR analysis of SOX2 mRNA expression in HOXA5 siRNA-transfected LHK2 cells. Asterisks indicated significant differences. *P values. Paired t-test. (G) Quantitative real-time PCR analysis of SOX2 mRNA expression in si Control-TSA(–) cells, si Control-TSA(+) cells, si HOXA5-TSA(–) cells and si HOXA5-TSA(+) cells. Asterisks indicated significant differences. *P, **P, ***P values. Paired t-test. (H) Tumor growth curves of siHOXA5 and siSOX2 transfected cells. derived from LHK2 cells injected in NOD/SCID mice, and representative views of mouse tumors. Each value is the mean tumor volume ± SD. *P, **P, ***P values. Paired t-test. (I) Binding of SOX2 promoter and HOXA5 promoter to acetylated histone shown by ChIP assay. β-actin was used as a positive control. Input: total Input DNA, ChIP IgG: normal rabbit IgG antibody, ChIP H3: acetyl-Histone H3 antibody. Right bar graph represents the relative mean grayvalue measured by ImageJ software (ChIP H3/Input).
To investigate whether HOXA5 induces the expression of SOX2, we performed a luciferase assay using luciferase DNA fused to the SOX2 promoter region. Co-transfection with HOXA5 induced significant luciferase activity compared to that in control cells (Figure 3B). To confirm the transcription activity of HOXA5, we transfected HOXA5 cDNA into LHK2 cells and investigated SOX2 expression by qRT-PCR. HOXA5 transfection was confirmed by qRT-PCR (Supplementary Figure S4H) and we found that HOXA5 transfection enhanced the expression of SOX2 (Figure 3C). The ratio of SP cells was increased by HOXA5 transfection (Figure 3D). Furthermore, we investigated whether treatment with TSA has an additional effect on SOX2 expression in HOXA5-transfected LHK2 cells. HOXA5 expression levels in empty vector-transfected TSA(–) cells, empty vector-transfected TSA(+) cells, HOXA5-transfected TSA(–) cells and HOXA5-transfected TSA(+) cells were confirmed by qRT-PCR (Supplementary Figure S4I). TSA treatment enhanced SOX2 expression in both HOXA5 non-transfected and transfected LHK2 cells, and HOXA5-transfected TSA(+) cells showed the highest SOX2 expression (Figure 3E). SP cells increased by HOXA5 transfection was cancelled by SOX2 knockdown using SOX2 siRNA (Figure 3D).
To confirm the SOX2 inducing role of HOXA5, a gene knockdown study using HOXA5-specific siRNA was performed. We designed HOXA5-specific siRNA and confirmed gene knockdown by qRT-PCR using HOXA5 siRNA-transfected LHK2 cells (Supplementary Figure S4J). SOX2 expression level was repressed by HOXA5 gene knockdown (Figure 3F). Furthermore, we investigated whether knockdown of HOXA5 repressed the induction of SOX2 by TSA treatment. The expression of HOXA5 in control siRNA-transfected TSA(–) cells, control siRNA-transfected TSA(+) cells, HOXA5 siRNA-transfected TSA(–) cells and HOXA5 siRNA-transfected TSA(+) cells was examined by qRT-PCR (Supplementary Figure S4K). And we found that HOXA5 gene knockdown also cancelled the SOX2 expression induced by TSA treatment (Figure 3G). HOXA5 knockdown by siRNA suppressed the sphere-forming ability of LHK2 cells (Supplementary Figure S4L). Furthermore, HOXA5 knockdown by siRNA suppressed the tumorigenicity of LHK2 cells as well as SOX2 knockdown (Figure 3H). To investigate whether SOX2 and HOXA5 promoter regions bind to acetylated histone, we performed a ChIP-PCR assay using an acetyl-Histone H3 antibody. Consistent with the results of qRT-PCR, the DNA fragments including each of the SOX2 and HOXA5 promoter regions encompassing the acetylated histone were pulled down more in the cells treated with TSA (Figure 3I).
HOXA5 represses expression of the tumor suppressor gene TP53 in lung cancer
It was reported that the tumor suppressor gene TP53 is activated by HDAC inhibitors [24–26], and if the status of TP53 is wild type, it will have a suppressive effect on tumor progression [27–30]. The status of TP53 in LHK2 cells was in fact analyzed by a next-generation DNA sequencer, and it was found that LHK2 cells have wild-type TP53 (data not shown). In addition, it was reported that HOXA5 promotes TP53 expression in breast cancer and other cancers [31–33], and TP53 was reported to suppress cancer stemness [34]. We thus investigated whether HOXA5 promotes TP53 expression in lung cancer cells. We transfected HOXA5 cDNA in lung cancer cell lines (LHK2, A549 and Sq-1 cells) and breast cancer cell lines (MCF7 cells) (Figure 4A). It was found that A549 and Sq-1 cells also have wild-type TP53 (data not shown). We analyzed TP53 expression levels by qRT-PCR. The expression of TP53 was significantly repressed by HOXA5 cDNA transfection in lung cancer cell line, whereas the expression of TP53 was significantly enhanced by HOXA5 cDNA transfection in breast cancer cell line as reported previously (Figure 4B) [31]. Furthermore, treatment of LHK2 cells with TSA enhanced the expression of HOXA5 (Figures 3B and 4C) and repressed the expression of TP53 (Figure 4D).
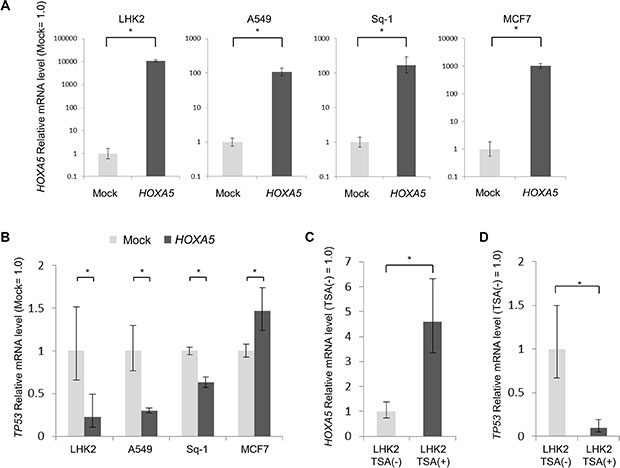
Figure 4: HOXA5 repress TP53 expression in lung cancer. (A) Quantitative real-time PCR analysis of HOXA5 mRNA expression in LHK2, A549, Sq-1 and MCF7 cells transfected with HOXA5. Empty vector-transfected cells were used for the control, which was set as 1.0. Data are expressed as means ± s.d. of relative values compared with empty vector-transfected cells. Asterisks indicated significant differences. *P < 0.05. t-test. (B) Quantitative real-time PCR analysis of TP53 mRNA expression in HOXA5-transfected cells (LHK2, A549, Sq-1 and MCF7 cells). Asterisks indicated significant differences. *P < 0.05. t-test. (C) (D) Quantitative real-time PCR analysis of HOXA5 and TP53 mRNA expression in LHK2 treated with TSA. Cells not treated with TSA were used for the control, which was set as 1.0. Data are expressed as means ± s.d. of relative values compared with nontreated cells. Asterisks indicated significant differences. *P < 0.05. t-test.
A previous study showed that the HOXA5 promoter region is methylated in breast cancer cells and that lack of HOXA5 expression may be one mechanism underlying the loss of TP53 expression [31]. In fact, MCF7 cells treated with the DNA methyltransferase inhibitor 5aza showed increased a significantly expression level of HOXA5 (Supplementary Figure S5). On the other hand, treatment with 5aza did not increase the expression level of TP53 in lung cancer cell lines (Supplementary Figure S5). The difference of epigenetic regulation in the HOXA5 promoter region may control the expression of TP53.
Repression of HDAC8 by oxidative stress is related to acquisition of stemness in lung cancer
We showed that lung cancer cells dedifferentiate into CSCs/CICs by epigenetics, and we also investigated the factor inducing the dedifferentiation. Lung cancer frequently develops in patients with chronic obstructive pulmonary disease (COPD), an inflammatory lung disease, and it has recently been reported that etiology of COPD is related to the expression of inflammatory genes induced by histone acetylation due to oxidative stress [35, 36]. We examined whether LHK2 cells acquire stemness by oxidative stress and increase the expression of HOXA5 and SOX2.
We confirmed oxidative stress by enhancement of the level of reactive oxygen species (ROS) using treatment with hydrogen peroxide (H2O2) for one hour in LHK2 cells. Each concentration of H2O2 increased the level of ROS (Supplementary Figure S6A). Many of the cells died with 10 mM H2O2 treatment, and following experiments were performed using 100 μM and 1 mM H2O2. First we confirmed that cells subjected to oxidative stress acquired chemoresistance ability as stemness. The results showed that LHK2 cells treated with 1 mM H2O2 had higher cell viability rates than those of control cells (Figure 5A). Furthermore, treatment with H2O2 increased the ratio of SP cells compared with that in control cells (Figure 5B).
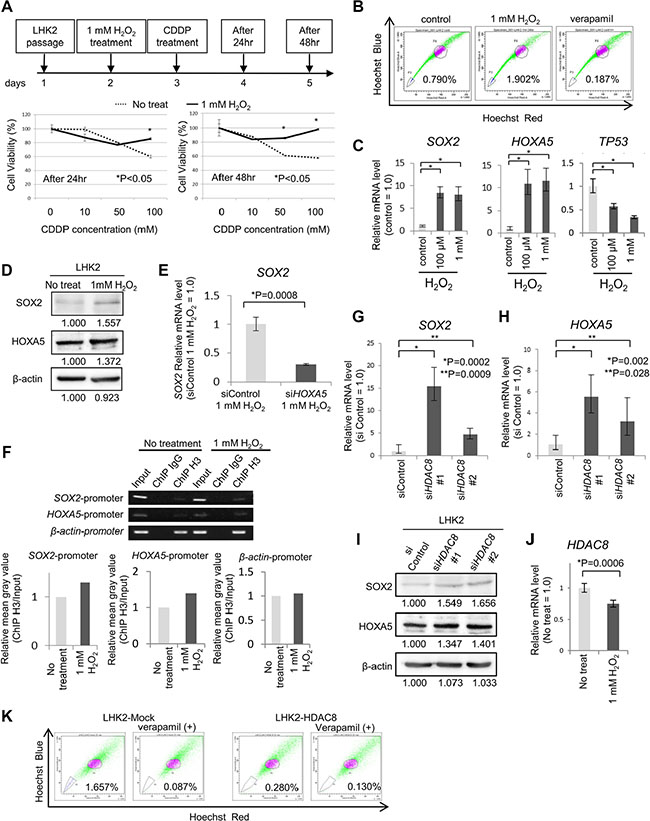
Figure 5: Repression of HDAC8 by oxidative stress may be related to acquisition of stemness. (A) Cell proliferation assays were performed in LHK2 cells treated with 1 mM H2O2 for 1 hr and cisplatin (CDDP) by using a Cell Counting Kit-8 and analyzed after 24 hr and 48 hr. (B) LHK2 cells were treated with 1 mM H2O2 for 1 hr and analyzed by SP assay after 24 hr. The percentage represents the ratio of SP cells. (C) Quantitative real-time PCR analysis of HOXA5, SOX2 and TP53 mRNA expression in LHK2 cells treated with 100 μM for 1 hr and 1 mM H2O2 for 1 hr. Non-treated cells were used for the control, which was set as 1.0. Data are expressed as means ± s.d. of relative values compared with nontreated cells. Asterisks indicated significant differences. *P < 0.05. t-test. (D) Expression of SOX2 and HOXA5 protein in LHK2 cells not treated with H2O2 (No treat) and 1 mM H2O2-treated cells (1 mM H2O2) confirmed by Western blotting with an anti-SOX2 antibody and an anti-HOXA5 antibody. (E) Quantitative real-time RT-PCR analysis of SOX2 mRNA expression in HOXA5 siRNA-transfected cells treated with 1 mM H2O2 for 1 hr. Control siRNA-transfected cells treated with 1 mM H2O2 cells were used for the control, which was set as 1.0. Data are expressed as means ± s.d. of relative values compared with control cells. Asterisks indicated significant differences. *P values. t-test. (F) Binding of SOX2 promoter and HOXA5 promoter to acetylated histone shown by ChIP assay. β-actin was used as a positive control. Input: total Input DNA, ChIP IgG: normal rabbit IgG antibody, ChIP H3: acetyl-Histone H3 antibody. Right bar graph represents the relative mean gray value measured by ImageJ software (ChIP H3/Input). (G) Quantitative real-time PCR analysis of SOX2 mRNA expression in HDAC8 siRNA-transfected LHK2 cells. Asterisks indicated significant differences. *P, **P values. t-test. (H) Quantitative real-time PCR analysis of HOXA5 mRNA expression in HDAC8 siRNA-transfected LHK2 cells. Asterisks indicated significant differences. *P, **P values. t-test. (I) Expression of SOX2 and HOXA5 protein in HDAC8 siRNA-transfected LHK2 cells confirmed by Western blotting with an anti-SOX2 antibody and an anti-HOXA5 antibody. (J) Quantitative real-time PCR analysis of HDAC8 mRNA expression in LHK2 cells treated with 1 mM H2O2 for 1 hr. Asterisks indicated significant differences. *P values. t-test. (K) SP analysis of HDAC8 overexpressed LHK2 cells. The percentage represents the ratio of SP cells.
We then examined SOX2 and HOXA5 expression levels in LHK2 cells treated with H2O2. Each concentration of H2O2 increased the expression of SOX2 and HOXA5 (Figure 5C and 5D). The protein expressions of SOX2 and HOXA5 is smaller than those in transcripts. This might depend on the efficacy of translation and the stability of proteins. TP53 expression was decreased by H2O2 (Figure 5C). Furthermore, we investigated whether knockdown of HOXA5 repressed the induction of SOX2 by oxidative stress. We examined the HOXA5 expression level in HOXA5 knockdown cells treated with 1 mM H2O2 by qRT-PCR (Supplementary Figure S6B), and we found that knockdown of HOXA5 cancelled the induction of SOX2 expression by treatment with 1 mM H2O2 (Figure 5E).
To determine whether SOX2 and HOXA5 promoter regions bind to acetylated histone in cells subjected to oxidative stress, we performed a ChIP-PCR assay using an acetyl-Histone H3 antibody. The results showed that DNA fragments including each of the SOX2 and HOXA5 promoter regions encompassing the acetylated histone were pulled down more in cells treated with 1 mM H2O2 (Figure 5F).
We then investigated which type of class I HDAC is related to the expression of SOX2. Class I HDACs consist of HDAC1, 2, 3 and 8, and we performed gene knockdown using HDAC-specific siRNAs. We designed two different specific siRNAs in HDAC1, HDAC2, HDAC3 and HDAC8 and confirmed gene knockdown by qRT-PCR using LHK2 cells transfected with each of the HDAC siRNAs (Supplementary Figure S6C). Knockdown of HDAC1 and HDAC2 did not change the expression of SOX2 (Supplementary Figure S6D). One of the HDAC3-specific siRNAs increased the expression of SOX2, but the other did not (Supplementary Figure S6D). LHK2 cells transfected with two different HDAC8-specific siRNAs showed higher expression levels of SOX2 than those in control siRNA-transfected cells (Figure 5G and 5I). On the other hand, HDAC8 overexpression decreased the SP cells in LHK2 cells (Figure 5K). HOXA5 expression was also increased by HDAC8 knockdown (Figure 5H and 5I). HDAC8 siRNA#1 and siRNA#2 decreased HDAC8 at similar levels; however, the expression levels of SOX2 and HOXA5 were different. This might be off-target effect of siRNAs. Finally, we examined the expression of class I HDACs by treatment with oxidative stress. Only HDAC8 was significantly repressed by treatment with 1 mM H2O2, suggesting that repression of HDAC8 by oxidative stress is the initial event for acquiring lung cancer stemness (Figure 5J and Supplementary Figure S6E).
DISCUSSION
Cancer is composed of heterogeneous subpopulations with regard to morphology and function. The ‘cancer stem cell model’ and ‘clonal evolution model’ were two major models to explain the heterogeneity of cancer [8, 37–39]. Recent studies have revealed that some differentiated non-CSCs/CICs can re-obtain a CSCs/CICs phenotype by various stimulations including microenvironment, and a ‘dynamic CSC model’ has been proposed to explain the plasticity of non-CSCs/CICs [40]. In this study, we analyzed the differentiation status and dedifferentiation status of human lung cancer cells at the single cell level and showed that some lung differentiated MP cells dedifferentiate into CSCs/CICs-like SP cells in short-term culture with relative high expression level of SOX2. Our data suggest that human lung cancer cells have plasticity by which lung differentiated non-CSCs/CICs can dedifferentiate into CSCs/CICs in vitro. Thus, the ‘dynamic CSC model’ might be a valid model for lung cancers.
Chaffer et al. showed that human mammary normal and neoplastic non-stem cells can convert to stem-like cells, suggesting plasticity of mammary epithelial cells [14]. Previous studies have shown that microenvironments including transforming growth factor-β (TGF-β) and hepatocyte growth factor (HGF) have roles in the plasticity of breast cancer cells and colon cancer cells, respectively [17, 41]. In our study, we showed that oxidative stress might be a mechanism by which dedifferentiation of non-CSCs/CICs is induced. Cigarette smoking is one of the risk factors of lung cancer [42], and cigarette smoke includes oxidants with other chemical carcinogens. Cigarette smoke thus might be one source for oxidative stress for carcinogenesis in smokers. Inflammation induced by respiratory diseases including COPD and Interstitial pneumonia is another risk factor of lung cancer [43, 44]. Immune cells including neutrophils and macrophages produce reactive oxygen species (ROS). Thus, ROS produced by immune cells might be another source of oxidative stress.
In a previous study, we showed that SOX2 is expressed in lung CSCs/CICs and that SOX2 has an essential role in the maintenance of lung CSCs/CICs [18]. We showed that the expression of SOX2 is upregulated more in dedifferentiated CSCs/CICs derived from non-CSCs/CICs than in non-CSCs/CICs, indicating that SOX2 might be a responsible key molecule in the dedifferentiation of lung cancer cells. Activation of epithelial-mesenchymal transition (EMT) and Wnt/β-Catenin signaling have been shown to have roles in dedifferentiation of breast cancer and colon cancer, respectively [16, 17, 41]. Thus, different molecular mechanisms are involved in dedifferentiation in different types of cancers.
SOX2 and its partner POU5F1 (OCT3/4) complex has transcriptional activity of SOX2 itself in ES cells [45]. However, the transcription factors of SOX2 in cancer cells have remained unknown. In this study, we investigated the gene expression mechanisms of SOX2 and found by a luciferase assay that HOXA5 is one of the transcription factors to induce SOX2 expression. Treatment by TSA increased the expression of SOX2 under overexpression of HOXA5. Thus another transcription factor derived by TSA or histone acetylation status of SOX2 promoter may also involved in the expression of SOX2. However, HOXA5 was reported to be a tumor-suppressor gene that can induce a TP53 tumor-suppressor gene [31] and that it is related to lung development [46]. Previous studies showed that the promoter region of HOXA5 is inactivated by DNA methylation in breast and lung cancer cells [31, 47]. However, our results showed that HOAX5 transcription in lung cancer cells was not induced by 5aza treatment, whereas HOAX5 transcription was induced by 5aza treatment in breast cancer cells. These observations indicate that the methylation status of the HOAX5 promoter region is variable and it might depend on several conditions. We found that the HOXA5 promoter was regulated by acetylation of H3 that can be induced by repression of HDAC8 expression by oxidative stress. Thus, oxidative stress might play a role in induction of lung CSCs/CICs by histone acetylation and inducing HOXA5 expression followed by SOX2 expression and TP53 repression. Lungs are always exposed to oxidative stress, and oxidative stress was shown to induce histone acetylation by repression of HDAC2 in the lung adenocarcinoma cell line A549 [35, 36]. These observations indicate that oxidative stress can modulate epigenetic gene expression regulation by repressing HDACs; however, the molecular mechanisms are still elusive.
Recently, HDAC inhibitors have been expected to have effects as adjuvants for tumors [48–50] and it is thought that TP53 is related to the effect [51]. Since it had been reported that TP53 is induced by HOXA5 [31], we investigated the interaction between them. Interestedly, HOXA5 induced wild-type TP53 in breast cancer cells as described previously. On the other hand, HOXA5 repressed wild-type TP53 expression in lung cancer cells. The molecular mechanisms by which HOXA5 showed different functions are still unknown; however, repression of TP53 might be one of the mechanisms by which CSCs/CICs are induced by HOXA5 in lung cancer cells. SOX2 is expressed in some normal adult tissues including brain [18], however, HOXA5 is only upregulated under the induction of SOX2 expression. These observations suggest that HOXA5 can be a reasonable target for CSC/CIC-targeting therapy.
Taken together, lung non-CSCs/CICs can dedifferentiate into lung CSCs/CICs by histone acetylation. Histone acetylation induces the expression of HOXA5, resulting in repression of TP53 expression and induction of SOX2 expression, which is responsible for the maintenance of CSCs/CICs. Histone acetylation can be induced by repression of HDAC8 expression by oxidative stress. These observations suggest that lung cancer cells can be dedifferentiated by oxidative stress and that the transcription factor HOXA5 has a critical role and HOXA5 can be a reasonable molecular target of lung CSC/CIC-targeting therapy (Figure 6).
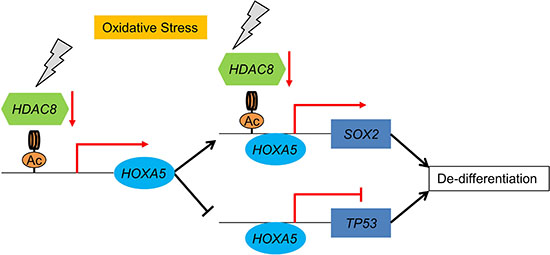
Figure 6: Schematic summary of lung cancer cell dedifferentiation model. Oxidative stress represses the expression of HDAC8, resulting in an increase in the acetylation of histone H3 of HOXA5 and SOX2 promoter regions. Upregulated HOXA5 induces the transcription of SOX2 and represses the transcription of TP53.
MATERIALS AND METHODS
Ethics statement
Mice were maintained and experimented on in accordance with the guidelines after approval by the Committee of Sapporo Medical University (No.10-032).
Side population (SP) assay
Side population (SP) cells were isolated as described previously using Hoechst 33342 dye (Lonza, Basel, Switzerland) with some modifications [19, 20]. Briefly, cells were resuspended at 1 × 106/mL in pre-warmed DMEM supplemented with 5% FBS. Hoechst 33342 dye was added at a final concentration of 2.5 μg/mL in the presence or absence of verapamil (75 μM; Sigma-Aldrich) and the cells were incubated at 37°C for 60 min or 90 min with intermittent shaking. Analyses and sorting were performed with a FACSAria II cell sorter (Becton Dickinson). The Hoechst33342 dye was excited at 357 nm and its fluorescence was analyzed using dual wave-lengths (blue, 402–446 nm; red, 650–670 nm).
Method for establishing SP clone cells and MP clone cells
SP cells and MP cells were isolated from LHK2 cells as described above and were plated at a single cell per well in a 96-well plate. Sorted single cells were cultured in DMEM supplemented with 10% FBS at 37°C in a humidified 5% CO2 atmosphere, and SP clone cells and MP clone cells were obtained after one week. We also established SP clone cells and MP clone cells derived from one population of MP clone cells by the same method.
Quantitative real-time PCR analysis
Quantitative real-time PCR was performed using a StepOne and StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. Primers and probes were designed by the manufacturer (TaqMan gene expression assays; Applied Biosystems). Thermal cycling was performed using 40 or 45 cycles of 95°C for 15 sec followed by 60°C for 1 min. Each experiment was done in triplicate, with normalization to the GAPDH gene as an internal control.
Methods for treatment with 5′–aza-2′- deoxycytidine, valproic acid and Trichostatin A
Cells were treated with 5′-aza-2′-deoxycytidine (5aza) (5 μM; Sigma-Aldrich), valproic acid (VPA) (4 mM; Sigma-Aldrich) and Trichostatin A (TSA) (100 nM; Sigma-Aldrich) according to the manufacturer’s protocol.
Cell transfection
An expression vector of pcDNA3.1 harboring HOXA5 cDNA was used for transfection into LHK2 cells. Transfection of cells was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. For stable transfection of HOXA5 and HDAC8 genes, a retrovirus vector pMXs-puro was used as described previously [21].
Luciferase assay
The SOX2 promoter connected downstream to the Luciferase gene was purchased from SwitchGear Genomics, a pCMV-LacZ vector was purchased from Clontech, and an expression vector of pcDNA3.1 harboring HOXA5 cDNA was established in our laboratory. Luciferase assay was performed using Luciferase Assay System (Promega) according to the manufacturer’s protocol, and β-galactosidase assay as an internal control was performed using a High Sensitivity β-Galactosidase Assay Kit (Agilent Technologies) according to the manufacturer’s protocol.
Small interfering RNA transfection
HOXA5 small interfering RNA (siRNA) was designed and synthesized using the BLOCK-it RNAi designer system (Life Technologies). The oligonucleotide encoding HOXA5 siRNA was 5′- AUUGCUCGCUCACG GAACUAUGAUC -3′. Cells were seeded at 50% confluence, and transfections were carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in Opti-MEM according to the manufacturer’s protocol.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the Acetyl-Histone H3 Immunoprecipitation (ChIP) Assay Kit (Upstate) according to the manufacturer’s protocol. In brief, 1 × 106 TSA-treated and untreated cells, and bulk and 1 mM H2O2 for 1 hr treated cells were cross-linked by adding formaldehyde directly to the culture medium. Cells were harvested and sonicated to shear DNA to lengths between 100 and 200 bp. After centrifuging samples for 10 min at 13,000 rpm at 4°C, the supernatant was pre-cleared with 80 μl of Salmon Sperm DNA/Protein A-Agarose-50% Slurry for 30 min at 4°C with agitation. Two μl of Acetyl-Histone H3 antibody was then added to the supernatant fraction for incubation overnight at 4°C with rotation. Then 60 μl of Salmon Sperm DNA/Protein A-Agarose was added to collect the antibody-histone complex. The protein A-agarose-antibody-histone complex was extensively washed for 5 min as suggested and then eluted with elution buffer and heated at 65°C for 6 h to reverse histone-DNA crosslinks. The DNA was recovered by phenol/chloroform extraction and ethanol precipitation. PCR was performed using two pairs of primers (SOX2 promoter: 5′- CAATGACACACCAACTCCTGC -3′ and 5′- CACACGCCTTTTCGAAGGAA -3′ ; HOXA5 promoter: 5′- TCAAGGAGAACCCTCCGACT -3′ and 5′- TGTTTCTCCAAGGCGAGGTC -3′ ; β-Actin: 5′- CCAGAGCAAGAGAGGCATCC -3′ and 5′- AGAGTCCTACGGAAAACGGC -3′).
Xenograft transplantation in NOD/SCID mice
LHK2 cells traesfected with control siRNA, HOXA5 siRNA and SOX2 siRNA were resuspended at concentrations of 1 × 103 cells in phosphate buffered saline and Matrigel (BD Biosciences) mixture (1:1), and were injected subcutaneously into the right and left mid back areas of anesthetized non-obese diabetic/severe combined immunodeficient (NOD/SCID) female mice (Charles River Laboratory Japan, Yokohama, Japan) at the age of 4–6 weeks. Tumor growth was monitored weekly, and tumor volume was calculated by XY2 / 2 (X = long axis, Y = short axis).
Cell proliferation assay
Cell proliferation assays of LHK2 cells were performed by using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Cells were plated in 96-well plates at 1 × 104 cells per well and cultured in DMEM (SIGMA, Ishikari, Japan) supplemented with 10% FBS (Life Technologies Japan, Tokyo, Japan) at 37°C in a humidified 5% CO2 atmosphere. The next day, LHK2 cells were treated with 1 mM H2O2 or not treated, and on the third day, they were treated with cisplatin (CDDP) at different concentrations. The cell numbers in triplicate wells were measured as the absorbance (450 nm) of reduced WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt).
ACKNOWLEDGMENTS AND FUNDING
This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to N. S.), program for developing the supporting system for upgrading education and research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to N. S.) and Takeda Science Foundation (to Y. H.), Sagawa Foundation for Promotion of Cancer Research (to Y.H.), Suharakinenzaidan Co., Ltd. (to Y.H.) and Kobayashi foundation for cancer research (to Y.H.). This study was supported in part by Grants-in-Aid for Regional R&D Proposal-Based Program from Northern Advancement Center for Science & Technology of Hokkaido Japan (to Y.H. and T.T.) This research is supported by the Project for Cancer Research And Therapeutic Evolution (P-CREATE) from Japan Agency for Medical Research and development, AMED.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer epidemiology, biomarkers & prevention. 2010; 19:1893–1907.
2. Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, Lin C, Leach C, Cannady RS, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012; 62:220–241.
3. Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. Journal of clinical oncology. 2003; 21:2237–2246.
4. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–1500.
5. Cappuzzo F, Janne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi L, Roncalli M, Destro A, Incarbone M, Alloisio M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Annals of oncology. 2009; 20:298–304.
6. Hammerman PS, Janne PA, Johnson BE. Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Clinical cancer research. 2009; 15:7502–7509.
7. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008; 8:755–768.
8. Clevers H. The cancer stem cell: premises, promises and challenges. Nature medicine. 2011; 17:313–319.
9. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994; 367:645–648.
10. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature medicine. 1997; 3:730–737.
11. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100:3983–3988.
12. Park CY, Tseng D, Weissman IL. Cancer stem cell-directed therapies: recent data from the laboratory and clinic. Molecular therapy. 2009; 17:219–230.
13. Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012; 488:527–530.
14. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:7950–7955.
15. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011; 146:633–644.
16. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA, Weinberg RA. Poised Chromatin at the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity. Cell. 2013; 154:61–74.
17. Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nature cell biology. 2010; 12:468–476.
18. Nakatsugawa M, Takahashi A, Hirohashi Y, Torigoe T, Inoda S, Murase M, Asanuma H, Tamura Y, Morita R, Michifuri Y, Kondo T, Hasegawa T, Takahashi H, et al. SOX2 is overexpressed in stem-like cells of human lung adenocarcinoma and augments the tumorigenicity. Laboratory investigation. 2011; 91:1796–1804.
19. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. The Journal of experimental medicine. 1996; 183:1797–1806.
20. Inoda S, Hirohashi Y, Torigoe T, Morita R, Takahashi A, Asanuma H, Nakatsugawa M, Nishizawa S, Tamura Y, Tsuruma T, Terui T, Kondo T, Ishitani K, et al. Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells. The American journal of pathology. 2011; 178:1805–1813.
21. Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000; 7:1063–1066.
22. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007; 1:555–567.
23. Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes & development. 2003; 17:1253–1270.
24. Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000; 408:377–381.
25. Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, Sun J, Yu Y, Zhou W, Zheng Q, Wu M, Otterson GA, Zhu WG. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol Cell Biol. 2006; 26:2782–2790.
26. Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008; 133:612–626.
27. Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989; 57:1083–1093.
28. Pollock R, Lang A, Ge T, Sun D, Tan M, Yu D. Wild-type p53 and a p53 temperature-sensitive mutant suppress human soft tissue sarcoma by enhancing cell cycle control. Clinical cancer research. 1998; 4:1985–1994.
29. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006; 6:909–923.
30. Voltan R, Secchiero P, Ruozi B, Caruso L, Forni F, Palomba M, Zauli G, Vandelli MA. Nanoparticles loaded with Nutlin-3 display cytotoxicity towards p53(wild-type) JVM-2 but not towards p53(mutated) BJAB leukemic cells. Current medicinal chemistry. 2013; 20:2712–2722.
31. Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature. 2000; 405:974–978.
32. Asuthkar S, Stepanova V, Lebedeva T, Holterman AL, Estes N, Cines DB, Rao JS, Gondi CS. Multifunctional roles of urokinase plasminogen activator (uPA) in cancer stemness and chemoresistance of pancreatic cancer. Molecular biology of the cell. 2013; 24:2620–2632.
33. Yan W, Liu S, Xu E, Zhang J, Zhang Y, Chen X, Chen X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene. 2013; 32:599–609.
34. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014; 14:359–370.
35. Moodie FM, Marwick JA, Anderson CS, Szulakowski P, Biswas SK, Bauter MR, Kilty I, Rahman I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB journal. 2004; 18:1897–1899.
36. Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. The European respiratory journal. 2005; 25:552–563.
37. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976; 194:23–28.
38. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009; 138:822–829.
39. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012; 481:306–313.
40. Vermeulen L, de Sousa e Melo F, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. The lancet oncology. 2012; 13:e83–89.
41. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704–715.
42. Hecht SS. Tobacco smoke carcinogens and lung cancer. Journal of the National Cancer Institute. 1999; 91:1194–1210.
43. El-Zein RA, Young RP, Hopkins RJ, Etzel CJ. Genetic predisposition to chronic obstructive pulmonary disease and/or lung cancer: important considerations when evaluating risk. Cancer prevention research. 2012; 5:522–527.
44. Artinian V, Kvale PA. Cancer and interstitial lung disease. Current opinion in pulmonary medicine. 2004; 10:425–434.
45. Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. 2013; 140:4129–4144.
46. Boucherat O, Montaron S, Berube-Simard FA, Aubin J, Philippidou P, Wellik DM, Dasen JS, Jeannotte L. Partial functional redundancy between Hoxa5 and Hoxb5 paralog genes during lung morphogenesis. American journal of physiology Lung cellular and molecular physiology. 2013; 304:L817–830.
47. Liu XH, Lu KH, Wang KM, Sun M, Zhang EB, Yang JS, Yin DD, Liu ZL, Zhou J, Liu ZJ, De W, Wang ZX. MicroRNA-196a promotes non-small cell lung cancer cell proliferation and invasion through targeting HOXA5. BMC Cancer. 2012; 12:348.
48. Kawamata N, Chen J, Koeffler HP. Suberoylanilide hydroxamic acid (SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells. Blood. 2007; 110:2667–2673.
49. Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nature chemical biology. 2010; 6:238–243.
50. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nature reviews Drug discovery. 2012; 11:384–400.
51. Bhatt S, Ashlock BM, Toomey NL, Diaz LA, Mesri EA, Lossos IS, Ramos JC. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. The Journal of clinical investigation. 2013; 123:2616–2628.