
Introduction
Class-I phosphatidylinositol 3-kinase (PI3KI) is the enzyme responsible for converting phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3). In the nematode C. elegans, mutations inactivating AGE-1, the catalytic subunit of PI3KI, result in increased resistance to a variety of stresses and extension of lifespan [1, 2] by as much as 10-fold [3]. Although PI3K lies in the insulin-like receptor/IRS/PI3K/AKT/FOXO pathway, pleiotropic effects of age-1-null mutations greatly exceed those of other disruptions to that signaling cascade, suggesting the possible involvement of other PIP3-binding proteins [2, 3]. There are >200 human proteins with Pleckstrin Homology (PH) domains, augmented by additional phosphoinositide-binding-domain families (see below). However, very few proteins in these families have been shown to preferentially bind PIP3, and protein homology has proven insufficient to identify PIP3-specific binding [4]. In contrast, structural analysis has shown promise for predicting PH-domain binding preferences [5]. We used PIP3- and PIP2-affinity enrichment to compare proteins from isogenic strains that differ genetically in their ability to make PIP3, coupled to proteomic identification of the binding proteins. Molecular modeling of proteins that show preferential binding to PIP3 over PIP2 allows us to extend structural prediction beyond PH-domain proteins, with the potential for confirmation by functional testing.
Phosphorylated derivatives of phosphatidylinositol (phosphoinositides or PtdInsPs) are key components of multiple signal-transduction complexes that convey external signals to the interior of cells [6-8]. PtdInsPs play critical roles in diverse cellular processes including growth, development, reproduction, cancer and longevity [9-11]. Phosphoinositides comprise 10-20% of phospholipids, but only ~1% of membrane lipids [12]. Proliferating cancer cells have elevated PIP3 levels due to elevated activity of PI3KI [13] or reduced activity of the opposing phosphatase, PTEN [14, 15], best known as an anti-oncogene.
Proteins with reported specificity for PIP3 include the serine/threonine kinases AKT and PDK-1 [4]; Phospholipase C (PLC) isoforms [16]; atypical protein kinase C, aPKC (e.g., Mζ and ι / λ isoforms) [17]; cytohesins [4]; general receptor for phosphoinositides 1, GRP1 [18]; kindlin3 [19]; IL-2 inducible T-cell kinase, ITK [20]; Skap-hom [21]; and Arf GTPase-activating protein 1 [4, 22]. Protein domains reported to confer PIP3-specific binding in some proteins (although based on quite variable evidence) include pleckstrin homology (PH) domains [4], Phox-homology (PX) and FYVE domains [23, 24], P2X domains [25], and the SYLF domain [26].
While disruption of insulin and insulin-like signaling (IIS) pathways has been shown to extend lifespan in diverse species, the elimination of class-I PI3K confers at least 4-fold greater life extension than any other IIS mutation [3]. The basis for this heightened dependence on PI3KI remains unresolved, but may be related to the ability of PI3KI mutation to reduce protein aggregation [27], which accompanies normal aging but is elevated and neurotoxic in most or all neurodegenerative diseases. IIS, and in particular PI3KI, modulate neuronal processes including learning and neuron survival [28, 29]. In the current study we identified the PIP3-binding proteins from C. elegans and showed that some are involved in age-related traits including oxidative stress resistance and longevity in normal worms, as well as protein aggregation and associated functional impairment in C. elegans models of neurodegeneration-associated proteinopathy.
Results
PI3KI contributes to protein aggregation in diverse nematode models
Components of the IIS pathway, including PI3KI, have been implicated in diverse neuropathologies including Alzheimer’s disease [30-32]. PI3KI is a key mediator of protein aggregation [27] and the unfolded protein response [33], leading us to test whether its knockdown (by RNAi targeting the age-1 gene that encodes the PI3KI catalytic subunit) would rescue nematode models of protein aggregation. In adult C. elegans with muscle expression of a Q40::YFP transgene, age-1 knockdown reduced the number of fluorescent aggregates by >35% (Figure 1A; P < 10 -4). Moreover, in worms expressing human Aβ1-42 in muscle, amyloid-induced paralysis declined 46% after age-1 knockdown (Figure 1B; P = 0.02).
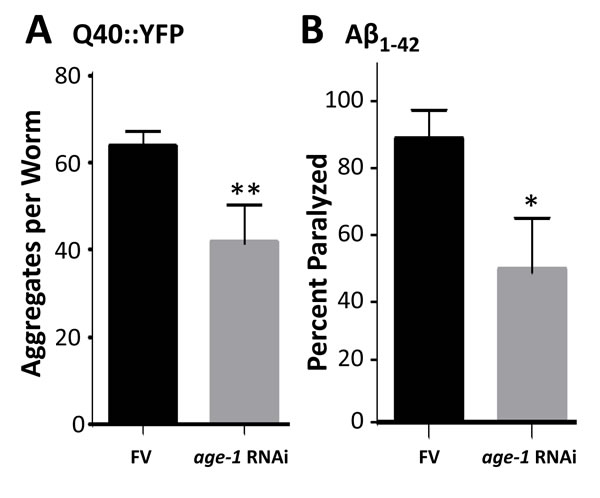
Figure 1: Knock-down of age-1 reduces Q40::YFP aggregates and protects against paralysis in worms expressing Aβ1-42 in body-wall muscle. A. assays of aggregate count in strain AM141 at adult day 4. B. assays of paralysis in strain CL4176 48 hr after induction of Aβ1-42. *P < 0.02; **P < 0.0001.
PIP3 deficiency reduces the yield of membrane proteins and especially of membrane-associated PIP3-binding proteins
The two fatty-acid chains of PIP3 are embedded in the inner plasma membrane, whereas PIP3-binding domains such as PH bind the phosphorylated inositol ring that projects into the cytoplasm. In this way, key signaling proteins such as AKT, PDK-1, PLCs, and aPKC are tethered to the inner membrane surface, where they are in proximity to other signaling kinases via clustering [34]. We used PIP3-coated agarose beads to isolate PIP3-binding proteins from C. elegans membranes, in which they are expected to be enriched. Based on staining of electrophoresed proteins, most but not all membrane proteins isolated from wild-type (N2) adults were less abundant in age-1-null mutant adults (Figure 2, solid arrows). Many protein bands, not necessarily the same ones, increased in abundance in worms fed a diet supplemented with exogenous PIP3 (open arrows, Figure 2).
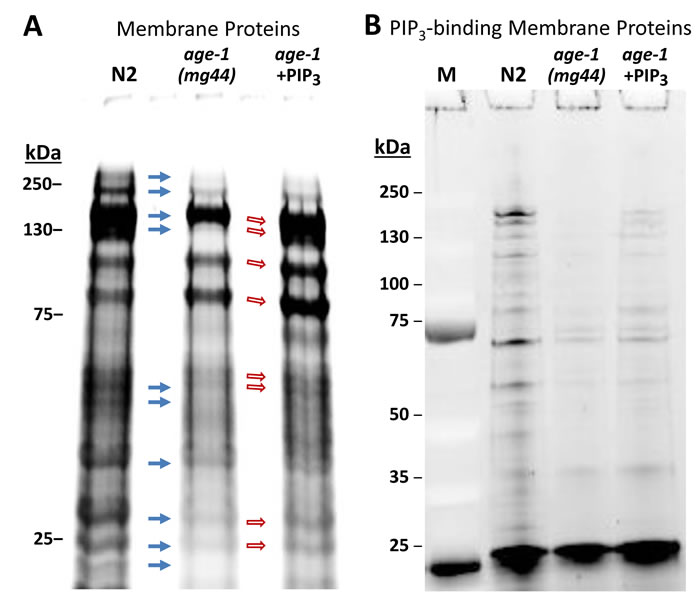
Figure 2: A strong nonsense mutation in the age-1 gene (allele mg44) reduces the recovery of many membrane proteins relative to wild-type controls, whereas feeding PIP3 to worms restores some bands. Polyacrylamide/SDS gels, stained with SYPRO Ruby after electrophoresis, show A. isolated membrane proteins, and B. proteins recovered from PIP3-coated beads, bound after isolation of membrane proteins as in A.
Gels like those of Figure 2 were sliced and analyzed by high-resolution proteomics to identify the proteins in each lane. Complete spectral counts are listed in Supplementary Data, Table S1, and summarized as Venn diagrams in Figure 3A. Of the 708 membrane proteins identified from N2, 632 (89%) were also seen in age-1(mg44) F2 adults lacking active PI3KI and having no detectable PIP3, so only 11% of these proteins may be membrane-associated via PIP3 tethering. Feeding PIP3 to PI3K-null worms restored 40 proteins that were identified in N2 (5.6%). Considering just those membrane proteins that bind PIP3 far more than PIP2 based on relative spectral counts (Figure 3B), 560 proteins from N2 adults met these criteria but just 286 of those (51%) were also identified in very long-lived age-1(mg44) F2 adults. Feeding PIP3 restored 81 proteins found in N2 (15%), or 30% of the 274 N2-specific proteins that might have been rescued.
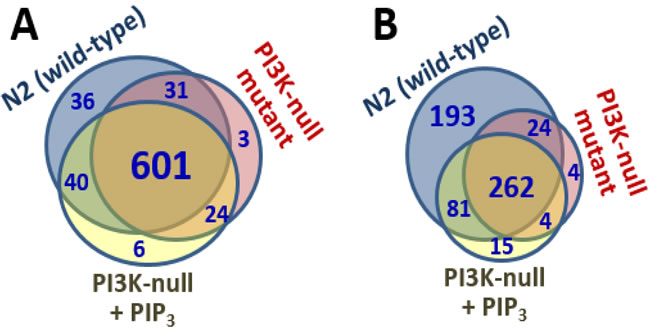
Figure 3: Venn diagrams. Indicate numbers of proteins positively identified in each membrane fraction (A), or in the subset of those proteins that bound PIP3>>PIP2 (B).
Proteins that preferentially bind PIP3
In Table 1, results are compiled from 3 typical experiments (of 5 that were run). The table lists many proteins that consistently preferred PIP3 over PIP2 binding (Experiments 1 and 2) and/or were greatly depleted in age-1(mg44) worms lacking active PI3KI and hence PIP3 (Experiment 3). Noteworthy differences are highlighted in yellow. Examples include AKT-1, a Ser/Thr kinase known to contain a PIP3-specific PH domain [4], which strongly preferred binding to PIP3 over PIP2 but was not effectively recovered from membranes of well-fed worms; muscle M-line assembly protein (UNC-89) and both nematode 14-3-3 proteins (PAR-5 and FTT-2), all reported to contain PH domains of uncertain specificity [4]; disorganized muscle protein 1; a variety of vitellogenins, protein precursors of LDL proteins also related to ApoB-100; chaperonins HSP60, HSP70, HSP90, and 4 subunits of the T complex, important in protein folding and refolding; protein disulfide isomerase, involved in refolding oxidized proteins; V-type proton ATPase; 3 fatty-acid binding proteins; 14 ribosomal proteins; and 5 α (noncatalytic) subunits of the 26S proteasome, most of which are in the 20S core assembly.
Table 1: PIP3-binding proteins identified in at least two experiments, based on affinity-binding capture ratios (Experiments 1 and 2) or genetic evidence (Experiment 3B).
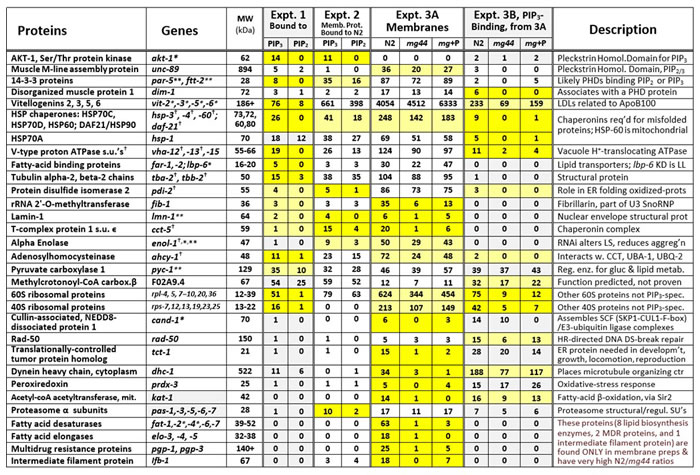
Related proteins that behaved similarly have been grouped together as indicated. Expt. 1: Worm (N2) proteins, recovered after affinity binding to PIP2- or PIP3-coated beads, were electrophoresed on polyacrylamide/SDS gels, and identified from trypsin-digested gel slices by LC-MS/MS proteomics. Expt. 2: Membrane proteins were isolated from wild-type worms (N2), and associated proteins were recovered using a detergent that dissociates protein complexes (unlike Expts. 1 and 3). Expt. 3A: Membrane proteins were isolated from wild-type worms (N2), a PI3K-null mutant (mg44) or mg44 adults fed PIP3. Expt. 3B: Proteins from 3A were bound to PIP3-beads, eluted, resolved by electrophoresis, and identified from trypsin-digested gel slices by LC-MS/MS proteomics. Spectral counts are shown, indicating the number of significant peptide identifications per protein, a crude measure of relative protein abundance. Deep yellow highlighting indicates ratios of ≥5; lighter yellow indicates suggestive differences. *RNAi knockdown extends lifespan; **RNAi reduces lifespan; †RNAi alters protein aggregation.
Gene ontology (GO) and pathway (KEGG) terms enriched among the proteins that preferentially bind PIP3 (Table 2) include Translation (enriched 7.4-fold, P < 10-64), Stress response (5.9-fold, P < 10-5), Mitochondria/respiration (5.6-fold, P < 10-5), Determination of adult lifespan and Aging (3.3-fold, P < 10-10), Lipid transport (2.9-fold, P < 0.003), Proteasome core complex (2.8-fold, P < 10-10), and Unfolded protein response (2.8-fold, P < 10-5).
Table 2: GO/Pathway analysis.
GO terms enriched for PIP3-specific binding proteins (total) |
|||
Term |
Count |
Fold Δ |
FDR |
Translation |
107 |
7.4 |
5E‒65 |
Protein biosynthesis |
40 |
2.4 |
2E‒43 |
Positive regulation of growth |
109 |
2.0 |
7E‒25 |
Adult life-span determination / Aging |
46 |
3.3 |
1E‒10 |
Proteasome core complex |
18 |
2.8 |
1E‒10 |
Proteasome component region PCI |
6 |
2.2 |
1E‒9 |
Stress response |
6 |
5.9 |
4E‒6 |
Unfolded protein response |
3 |
2.8 |
6E‒6 |
Mitochondria / Cellular respiration |
14 |
5.6 |
1E‒5 |
Lipid transport |
4 |
2.9 |
3E‒3 |
Functional-annotation enrichment (https:/david.ncifcrf.gov/) was analyzed for proteins recovered from PIP3-affinity beads and identified by LC-MS/MS. Count: total proteins per category. Fold Δ: term enrichment factor. FDR: False Discovery Rate (<0.05 is considered significant).
In silico modeling predicts preferential interaction with PIP3 over PIP2, for most proteins with higher observed affinity for PIP3
Molecular docking programs were used to estimate affinities for PIP3 vs. PIP2 of proteins empirically observed to prefer PIP3. The docking predictions are not expected to support all of the observed PIP3-specific candidate proteins, since retention on PIP3-coated beads could reflect either direct or indirect binding (e.g., via a complex). We began by retrieving crystallographic or NMR-based structures from the PDB database, for structure-defined orthologs of candidate proteins that had shown preferential affinity for PIP3 in multiple experiments. The corresponding C. elegans protein structures were then derived using molecular modeling with I-TASSER or MODELLER 9.13 (see Methods). Similarly, the NMR-based structures of PIP3 and PIP2 were refined in MODELLER, chiefly by truncating the fatty-acid chains to limit their contributions to interactions.
Docking of each protein structure was simulated with PIP2 and PIP3 separately by energy minimization, using AutoDock-Vina to calculate ΔGbinding (the change in Gibbs free energy on binding) for each docking interaction. Table 3 shows ΔGbinding for protein binding to PIP3 or PIP2 and the difference between them. That energy difference, ΔΔG = ΔG(PIP3)–ΔG(PIP2), indicates the binding preference for either phosphoinositide. Docking models that emphasize contact points (Figure 4A & 4B) illustrate the precise fit of PIP3 in PH domains of both human and nematode AKT proteins. Predicted ΔΔG values for 15 of 31 candidate proteins (48%) surpassed all 40 randomly chosen control proteins (Figure 4C; rank-order P < 3 x 10-4), and 16 of those (bold lines, Table 3) would be considered significant at an empirical threshold of P < 0.05. For all 16, ΔG(PIP3) was < ‒7.5, indicating relatively stable interactions.
Table 3: In silico interaction energies of PIP3-binding proteins with PIP3 vs. PIP2
Protein [Role] |
ΔGbinding for PIP3 (kCal/mol) |
ΔGbinding for PIP2 (kCal/mol) |
ΔΔG |
CYC-2.1 [one of 2 C. elegans cytochrome c proteins] |
‒10.6 |
‒6.5 |
‒4.1 |
RPL-21 [ribosome large s.u. protein 21, SH3 domain] |
‒13.6 |
‒9.8 |
‒3.8 |
DKC-1 [ortho. of human dyskerin, H/ACA RNP s.u.] |
‒10.4 |
‒7.3 |
‒3.1 |
FAT-2 (iso. A) [Δ12 FA desaturase, increases fluidity] |
‒9.7 |
‒7.2 |
‒2.5 |
NKB-1 [Na+/K+-transporting ATPase s.u.] |
‒9.2 |
‒7.2 |
‒2.0 |
CCT-1 [α subunit of T-complex chaperonin] |
‒9.0 |
‒7.1 |
‒2.0 |
AKT-1c [insulin-like signaling kinase] |
‒10.6 |
‒8.6 |
‒1.9 |
RAD-50a (C-term.) [part of homol. recomb. complex] |
‒9.1 |
‒7.3 |
‒1.8 |
IFE-1 (iso. A) [mRNA cap-binding protein eIF4E] |
‒10.1 |
‒8.3 |
‒1.7 |
EIF-1 [ortho. of euk. translation initiation factor EIF1] |
‒8.7 |
‒7.1 |
‒1.7 |
PAS-1 [α subunit 1 of 26S proteasome] |
‒11.7 |
‒10.0 |
‒1.7 |
TCT-1 [orthol. to human TPT1, tumor protein 1] |
‒9.4 |
‒7.8 |
‒1.6 |
SOD-2 [Fe++/Mn++ superoxide dismutase] |
‒7.9 |
‒6.3 |
‒1.6 |
MSP-78 [major sperm protein 78] |
‒8.8 |
‒7.3 |
‒1.6 |
HSP-6 [DnaK/Hsp70 family chaperone] |
‒7.5 |
‒6.2 |
‒1.4 |
KAT-1 [ketoacyl-coA thiolase, part of FA β-oxidation] |
‒8.9 |
‒7.7 |
‒1.2 |
HIPR-1 [ortho. to Huntingtin-interacting protein 1-r] |
‒6.3 |
‒5.7 |
‒0.6 |
CAND-1 [cullin-associated NEDD8-dissoc. protein 1] |
‒5.7 |
‒5.1 |
‒0.6 |
FAR-1 (iso. A) [fatty acid/retinol binding protein 1] |
‒10.9 |
‒10.5 |
‒0.4 |
MSP-77 [major sperm protein 77] |
‒8.7 |
‒8.4 |
‒0.3 |
SOD-3 [Fe++/Mn++ superoxide dismutase, orth. SOD1] |
‒6.8 |
‒6.7 |
‒0.1 |
CGH-1 [DEAD-box RNA helicase] |
‒8.3 |
‒8.4 |
0.1 |
SOD-1 (iso. A) [Cu++/Zn++ superoxide dismutase] |
‒6.2 |
‒6.7 |
0.5 |
MSP-3 [major sperm protein 3] |
‒8.1 |
‒8.7 |
0.6 |
PAS-2 [α subunit 2 of 26S proteasome] |
‒7.0 |
‒7.7 |
0.6 |
LBP-1 [lipid-binding protein 1] |
‒8.6 |
‒9.3 |
0.6 |
RPN-12 (iso. A) [19S proteasome, regulatory s.u. 12] |
‒5.5 |
‒6.4 |
0.9 |
SODH-1 [sorbitol dehydrogenase 1] |
‒8.1 |
‒9.2 |
1.1 |
IFB-1a_head [intermediate filament protein 1] |
‒5.9 |
‒7.2 |
1.3 |
RPN-2 [19S proteasome, regulatory subunit. 2] |
‒6.7 |
‒8.1 |
1.4 |
BCAT-1 [branched-chain aminotransferase 1] |
‒5.5 |
‒7.2 |
1.7 |
Control Proteins (top 10 proteins are shown, out of 40 taken at random from PDB) |
|||
1a3q |
‒9.8 |
‒8.6 |
‒1.2 |
1a3b 5th percentile ΔΔG threshold |
‒7.4 |
‒6.3 |
‒1.1 |
1a10 |
‒7.7 |
‒6.7 |
‒1.0 |
1agf 10th percentile ΔΔG threshold |
‒8.2 |
‒7.3 |
‒0.9 |
1a00 |
‒7.8 |
‒7.1 |
‒0.7 |
1a0z |
‒7.4 |
‒6.7 |
‒0.7 |
1ah1 |
‒5.9 |
‒5.3 |
‒0.6 |
1a17 |
‒6.0 |
‒5.4 |
‒0.6 |
1axa |
‒7.6 |
‒7.1 |
‒0.5 |
1a1m |
‒7.3 |
‒6.8 |
‒0.5 |
For each protein structure listed (obtained from PDB or derived as described), docking was simulated with PIP2 or PIP3 by energy minimization using AutoDock-Vina. ΔGbinding was calculated for each docking, and proteins were ranked by the difference between PIP3 and PIP2 binding energies: ΔΔG = ΔG(PIP3) –ΔG(PIP2). Control protein structures were taken at random from PDB for 40 proteins, of which the top-ranked 10 (based on ΔΔG) are shown.
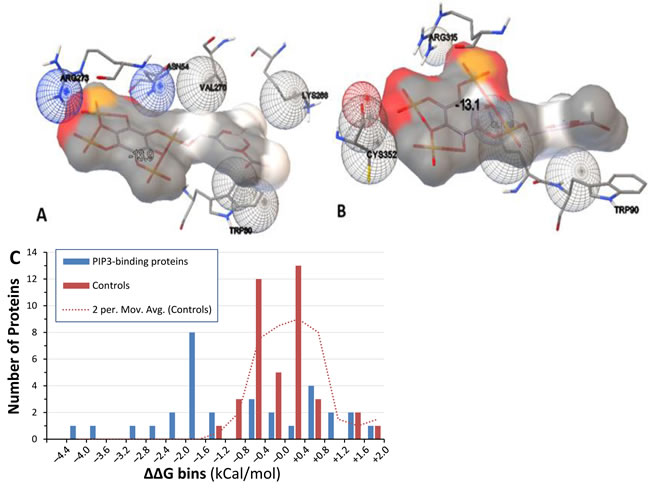
Figure 4: Docking models of PIP3 binding to proteins. A. & B. Hybrid models of PIP3 (grey) and contact residues of human AKT (A) with ΔGbinding of ‒13.9 kCal/mol, and nematode AKT-1 (B) with ΔGbinding of ‒13.1 kCal/mol. C. “bin plot” of ΔΔG values for PIP3-specific binding candidates (blue bars) and randomly chosen control proteins (red bars). Intervals are rightward inclusive (e.g. proteins are shown between –1.6 and –1.2 if ΔΔG lies between –1.59 and –1.2). Dotted line: smoothed control distribution, taken as a moving average of adjacent count pairs.
Knockdown of genes encoding PIP3-binding proteins enhances stress resistance
We selected 18 PIP3-binding proteins, based on function and inclusion in the Ahringer RNAi library [35], to ask whether they might contribute to the “multiple stress resistance” traits of C. elegans age-1-null mutants [36]. Knockdowns of 5 candidates (28%) significantly improved survival in hydrogen peroxide (Figure 5), an oxidative stress to which age-1(mg44) mutants are exceptionally resistant [3]; they are TCT-1, CAND-1, AKT-1, RAD-50 and FAT-2.
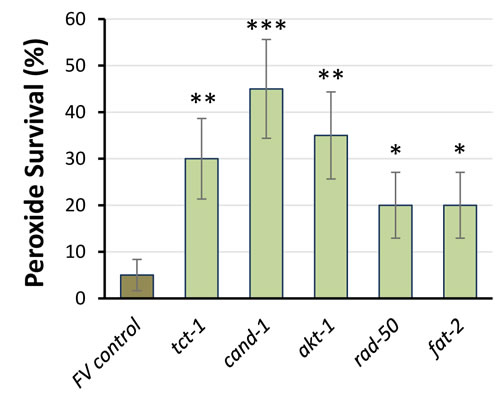
Figure 5: RNAi knockdown of PIP3-binding proteins improves peroxide survival. Worm survival ±SE is shown after 4h in 5-mM H2O2. RNAi groups differed significantly from controls (by 1-tailed Fisher exact test): *P < 0.05; **P < 0.005; ***P < 10-4 (each N = 40). Error bars indicate the standard error of a proportion.
PIP3-binding proteins influence protein aggregation in C. elegans model systems
The same 18 PIP3-binding candidates were tested to determine whether they contribute to protein aggregation. Strain CL4176 can be induced to express human Aβ1-42 in body-wall muscle [37]; it forms β-amyloid aggregates leading to paralysis soon after induction, or progressively with age if not induced [36]. RNAi knockdown of 5 genes (28%) encoding RAD-50, AKT-1, CAND-1, FAT-2 and DHC-1, reduced age-dependent paralysis in adult worms with “leaky” Aβ1-42 expression [36], shown at day 12 in Figure 6. Three of these knockdowns (rad-50, cand-1 and fat-2) also significantly blocked paralysis 48 h after induction of Aβ1-42 (data not shown), but we consider uninduced paralysis to be a more appropriate model of age-dependent protein aggregation.
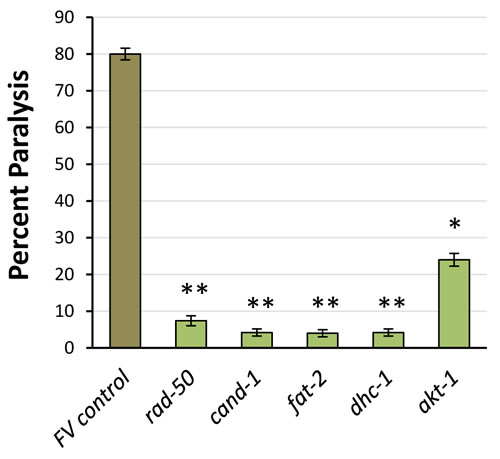
Figure 6: RNAi knockdown of PIP3-binding proteins reduces age-dependent paralysis from leaky muscle expression of Aβ1-42. Data show % paralyzed, ±SD, at 12 d post-hatch. RNAi groups differed significantly from controls (by 1-tailed Fisher exact test): akt-1, P < 10-4; all others, P < 10-7.
Muscle-specific expression of human α-synuclein (in strain NL5901), a model of Parkinson’s disease, was also assessed during adult aging. Significant reductions in the number of aggregates, at least as deep as that elicited by RNAi to age-1, were observed at 9 and 10 days post-hatch after knockdown of genes encoding 6 (33%) of 18 PIP3-binding proteins tested: RAD-50, FAT-2, TCT-1, PRDX-3, KAT-1, and PAS-6 (Figure 7). KD of ifb-1 appeared to increase aggregates, although without statistical significance.
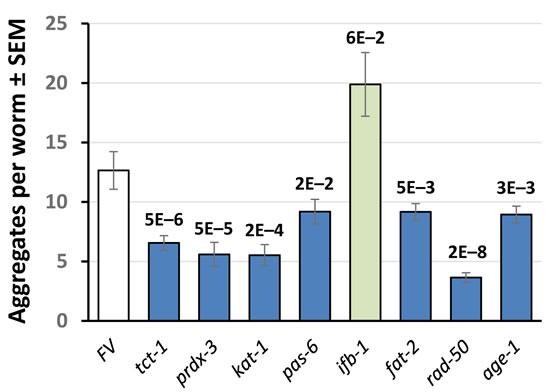
Figure 7: Aggregate counts for C. elegans day-9 adults expressing muscle α-synuclein. Adult worms were maintained for 9 days on bacteria expressing RNAi against the genes indicated, encoding PIP3-binding proteins. Error bars indicate ±SEM. Values above bars show significance of differences from Feeding Vector (FV) control, by heteroscedastic 2-tailed t-tests.
RNAi knockdowns of several PIP3-binding proteins confer multiple fitness benefits
Previous studies in C. elegans had reported lifespan extension upon knockdown for two of those 5 genes, akt-1 [38] and fat-2 [39]. We confirmed a significant (P < 0.05) life extension upon akt-1 knockdown (data not shown), and somewhat stronger effects of RNAi targeting cand-1 (**P < 10-4) or rad-50 (*P < 3x10-4) (Table 4, Figure 8), suggesting that these PIP3-binding proteins may also contribute to the unique longevity of age-1-null mutants that cannot form PIP3 [3]. Significant but less pronounced life extension was observed when RNAi was begun only at the L4 (late-larval) stage to avoid effects on development (Table 4, Expt. 4). However, no life extension was seen in a daf-16 mutant (Figure 8B), confirming that cand-1 and rad-50 act via the insulin/IGF-1 signaling pathway. Of 18 PIP3-binding candidates tested by RNA interference, 5 improved fitness by at least two measures, survival of peroxide stress and protection in protein-aggregation models (Table 5), and at least 4 extend lifespan.
Table 4: Survival data for PIP3-binding candidate knockdowns
Experiment 1 (N2) |
Expt. 2 (N2) |
Expt. 3 (N2) |
Expt. 4 (N2) |
||||||||||||
RNAi: |
none |
CAND-1 |
RAD-50 |
none |
CAND-1 |
none |
RAD-50 |
none |
RAD-50 |
||||||
Median |
18.5 |
23.5 |
23.5 |
23.5 |
28.5 |
22.5 |
25.5 |
22.5 |
22.5 |
||||||
Ratio |
― |
1.27 |
1.27 |
― |
1.21 |
― |
1.13 |
― |
1.0 |
||||||
Mean |
19.5 |
23.2 |
23.5 |
24.3 |
28.0 |
23.2 |
25.1 |
22.6 |
24.3 |
||||||
Ratio |
― |
1.19 |
1.20 |
― |
1.15 |
― |
1.10 |
― |
1.08 |
||||||
P < |
― |
3E–5 |
3E–5 |
― |
0.0001 |
― |
0.007 |
― |
0.008 |
||||||
Expt. 5 (N2) RNAi from L4 |
Experiment 6 (daf-16‒) |
Experiment 7 (daf-16‒) |
|||||||||||||
RNAi: |
none |
CAND-1 |
RAD-50 |
none |
CAND-1 |
RAD-50 |
none |
CAND-1 |
RAD-50 |
||||||
Median |
22.5 |
24.5 |
24.5 |
17.5 |
17.5 |
17.5 |
18.5 |
18.5 |
17.5 |
||||||
Ratio |
― |
1.09 |
1.09 |
― |
1.00 |
1.00 |
― |
1.00 |
0.95 |
||||||
Mean |
23.5 |
24.5 |
24.6 |
18.9 |
18.6 |
18.4 |
18.5 |
18.6 |
18.5 |
||||||
Ratio |
― |
1.04 |
1.05 |
― |
0.99 |
0.97 |
― |
1.00 |
1.00 |
||||||
P < |
― |
0.04 |
0.06 |
― |
0.6 |
0.4 |
― |
0.8 |
1.0 |
||||||
Median and Mean survivals are given in days post-hatch. Five independent experiments were performed in the wild-type N2-DRM strain, and two in strain SR814, created by outcrossing DR26 (daf-16(m26)) into N2-DRM. RNAi feeding began at hatch, or (Experiment 5) in late L4. Gehan-Wilcoxon
log-rank P values are shown.
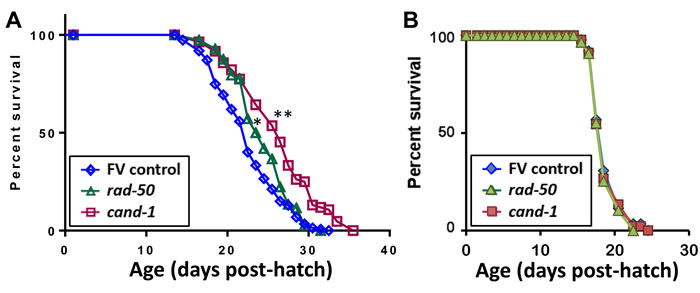
Figure 8: Survivals of C. elegans exposed from the late-larval (L4) stage, to RNAi targeting genes that encode two PIP3-binding proteins. Data were combined from 4 experiments with N2 wild-type worms (A) or from 2 experiments with daf-16(m26) mutant worms (B) exposed to empty feeding vector (FV) or RNAi targeting cand-1 or rad-50. Total numbers of uncensored/censored deaths included were (A) control, 148/7; cand-1 RNAi, 82/2; and rad-50 RNAi, 114/6; and (B) control, 88/2, cand-1 RNAi, 90/0; and rad-50 RNAi, 87/3. *Gehan-Wilcoxon log-rank P < 3E-4; **Gehan-Wilcoxon log-rank P < 1E-4.
Discussion
PIP3, the product of class-I PI3K, is normally embedded in eukaryotic cell plasma membranes where it is thought to contribute to multiple kinase-cascade signaling pathways. C. elegans mutants lacking the PI3KI catalytic subunit exhibit extreme longevity, improved stress resistance, delayed development, and reproductive defects [3]. We infer that PIP3 plays critical roles in these pleiotropic physiological traits by binding and recruiting signaling proteins. Those proteins, and the pathways they participate in, must confer developmental and reproductive benefits early in life, presumably through roles in anabolic metabolism and cell proliferation. Nonetheless, they appear to have deleterious long-term consequences so that their continued activity in post-gravid adults promotes aging.
Table 5: Multiple benefits of PIP3-binding candidate knockdowns
Gene |
H2O2 survival |
Aβ1–42 aggreg. |
α-synuclein aggregation |
Longevity effect (range) or reference |
induced aging |
||||
rad-50 |
↑5x |
↓3x ↓10x |
↓3.6x |
↑15% (8 – 20%) |
cand-1 |
↑11x |
↓5x ↓20x |
N.S. |
↑17% (15 – 19%) |
fat-2 |
↑5x |
↓4x ↓20x |
↓40% |
↑ [40] |
akt-1 |
↑9x |
N.S. ↓3x |
N.S. |
↑9%; [45] |
tct-1 |
↑7x |
N.S. N.S. |
↓ 2x |
N.S., not significant; grey shading, not tested here. Reference citations indicate published lifespan data.
Proteins that bind PIP3 are tethered to the membrane, dependent on PIP3 availability
We used a combined proteomic/genetic strategy to isolate and identify PIP3 binding proteins in C. elegans. Although any affinity-capture procedure can produce false positives, we set several criteria by which to evaluate candidate proteins identified in at least 3 independent experiments.
Preferential binding to PIP3 over PIP2
PIP3-specific binding was assessed by comparing the capture of any given protein based on affinity to each ligand. Many proteins showed differential binding, of which 10 (highlighted in bright yellow, Table 1/Expt. 1) met our arbitrary criteria of (i.) at least 5-fold higher binding to PIP3 than to PIP2 and (ii.) at least 5 peptide “hits”. Several caveats should be considered. The PIP3/PIP2 ratio in normal cells ranges from 0.001 to 0.02, suggesting that moderately high affinity ratios might not be sufficient to prevent binding to PIP2 due to its higher abundance in vivo. Moreover, affinity purification may not precisely mirror physiological dependence on ΔGbinding, since binding also depends on protein abundance. Protein levels are equal for the two PI ligands in lysates, but certainly differ among cell types in vivo, which might also vary in their PIP3 levels. Binding propensities in vivo can also be altered by additional factors such as competing ligands, other interacting proteins in a complex, and other hydrophobic or electrostatic features in the immediate vicinity of membrane-embedded PIP3.
Some proteins may show preferential affinity for PIP3 through indirect binding, in which the protein is part of a stable complex containing a PIP3-specific binding protein. Our data suggest that this may be quite common. We note that Experiments 1 and 2 in Table 1 differed in only one respect: protein complexes were disrupted in the second experiment, leading to loss of many “PIP3-specific” proteins that had ceased to be significantly differential. The value of candidate PIP3-binding proteins as potential pharmacologic targets, however, does not depend on whether membrane-tethering is direct or indirect.
Presence on cell membranes, dependent on biological availability of PIP3
We took advantage here of our previous studies demonstrating that PIP3 is essentially absent (below detectable limits, i.e. < 1% of normal levels) in second-generation homozygous age-1 mutants lacking active PI3KI or detectable PIP3 [2]. Thus we expected that genuine PIP3-binding proteins will be recovered from membranes of wild-type worms but not from extremely long-lived age-1(mg44) mutant worms. This expectation was met convincingly (>2-fold reduction and P < 0.05) by a subset of candidate proteins (Table 1, Expt. 3A): lamin-1, 2 chaperones (HSP60, HSP90), rRNA 2’-O-methyltransferase, T-complex subunit CCT-5, adenosylhomocysteinase, 11 ribosomal proteins, CAND-1, TCT-1, 5 fatty-acid desaturases and 2 elongases, 2 multidrug resistance proteins and an intermediate filament protein.
Many proteins are integral to membranes, and if these are in vast excess they might mask proteins that are recruited to membranes via tethering to PIP3. Thus it is reassuring that most PIP3-binding proteins that were affinity-isolated from the membrane fraction, although depleted in overall abundance by the two-step isolation, were further reduced in age-1(mg44) adult worms relative to N2 controls (Table 1, Experiment 3B). Apparent exceptions include pyruvate carboxylase 1, CAND-1, TCT-1, peroxiredoxin and proteasome α subunits.
Restoration of membrane tethering by exogenous PIP3
In principle, it should be possible to reverse all “age-1” traits in worms genetically deficient in PI3KI and hence in PIP3, by supplying exogenous PIP3, provided that it is taken up, is not degraded, and reaches all tissues. Of the proteins listed under (2.) above, fatty-acid desaturases and elongases showed essentially no reversal (≤3%) whereas all other proteins were partially reversed (17-80%) by PIP3 feeding.
This wide variation in efficacy of PIP3 supplementation appears paradoxical: If PIP3 is able to enter cells to reverse any of the age-1 phenotypes, should it not reverse all of them? Variable extents of rescue could result from our use of very short-acyl-chain PIP3, which may be insufficient to tether large proteins or complexes to the plasma membrane. A plausible alternative explanation is tissue heterogeneity of PIP3 uptake, since PIP3 added to the medium or the bacterial lawn would reach intestinal cells first, and unless it saturated the membranes of those cells it might not distribute any further. Our data appear inconsistent with this scenario, however, since the proteins least effectively “reverted” (restored to membrane fractions in age-1(mg44) F2 worms) by PIP3 feeding are fatty-acid desaturases and elongases, expected to be concentrated in the intestine since it serves as the lipid-storage organ of nematodes [40, 41].
Molecular modeling can predict proteins that preferentially bind to PIP3
Park et al. [5] used “machine learning” to define the amino-acid residues and positional constraints within PH domains that distinguished between proteins demonstrated previously (or, for the test group, demonstrated in that paper) to have binding specificity for particular PI moieties including PIP3. Apart from that study, no systematic attempt has been made to identify all proteins with PIP3-specific binding. Over the last few years, molecular modeling has proven itself capable of screening protein-drug and protein-protein interactions with ever-increasing reliability. Nevertheless, we were uncertain whether it would prove equal to the challenge posed by discriminant PIP3-binding.
Just over half of the 31 tested proteins that had been identified as strong PIP3-specific candidates substantially exceeded the minimal energetic requirements for avid and selective binding of PIP3, with ΔΔG values outside the range observed in randomly-chosen control proteins (Table 4; Figure 4C). We note that PIP3 would be considered a reasonable candidate ligand for all 31 proteins, given that their ΔG levels ranged from -5.5 to -13.6; protein ligands predicted previously by AutoDock agreed well with confirmed binders provided that ΔGbinding was < -4 [42]. Our results strongly support the premise that molecular modeling can predict substrate specificity for PtdInsP-binding proteins. The 15 proteins with unexceptional ΔΔG values may not selectively bind PIP3 themselves, but instead participate in larger protein complexes that include a PIP3-specific component. Such indirect tethering of complexes may be quite common, but the candidate proteins nevertheless remain drug targets of interest if they are components of complexes that require PIP3 or membrane localization to function.
PIP3-binding proteins are enriched for roles in translation, longevity and proteostasis
Interesting proteins that met multiple criteria for PIP3-specific binding include AKT signaling kinases (AKT-1, AKT-2); heat-shock chaperones (HSP60, HSP70s, and HSP90); T-complex protein CCT-1; 14-3-3 proteins (PAR-5, FTT-2); muscle M-line protein UNC-89; branched-chain amino-acyl transferase BCAT-1; ubiquitinylation/NEDD8 regulator CAND-1; RNA helicase CGH-1; proteasome subunits (PAS-1, PAS-2, RPN-7); 8 fatty-acid desaturases and elongases (FAT and ELO proteins), and 14 ribosomal proteins (8 RPL and 6 RPS).
Among these proteins are several previously reported to possess or associate with PIP3-binding domains (AKT [43]; 14-3-3 proteins [44]; DIM-1; UNC-89 [www.wormbase.org]), and a remarkably large number that were previously implicated in longevity and age-related diseases: AKTs [45]; HSPs [46]; 14-3-3 proteins [44]; fatty acid desaturases FAT-2 [39] and FAT-4 [40]; lipid-binding protein LBP-6 [47]; pyruvate carboxylase, PYC-1 [39]; α-enolase/ ENOL-1 [48]; and lamin-1 [49]. In addition, many have roles in proteostasis failure leading to protein aggregation, a process that is critical to lifespan regulation [36,50]: HSPs [46]; T-complex proteins [51]; V-type proton ATPase [51]; actin and tubulin chains [52]; disulfide isomerase [53]; DHC-1 [54]; adenosylhomocysteinase [51]; and proteasomes [36]. Dynein heavy chain 1 (DHC-1) is associated with neurodegenerative diseases and protein aggregation [54], and ketoacyl thiolase (KAT-1) is a conserved mitochondrial fatty-acid β-oxidase that delays aging, independent of known longevity pathways [www.wormbase.org].
The frequent recovery of ribosomal proteins, specific to PIP3 affinity and to worms with PI3KI activity, suggests that PIP3 may be involved in tethering ribosomes to rough endoplasmic reticulum. This conjecture is currently supported only by indirect evidence [55], but would account for the 7.4-fold GO enrichment for “translation” (Table 2).
PIP3-binding proteins contribute to diverse pathways mediating extreme age-1 traits
Although it was not a criterion for PIP3-specific binding, a goal of the current study was to assess whether any of the proteins downstream of PI3KI, if depleted by RNA interference, could confer some part of the beneficial survival traits displayed so strikingly by very-long-lived age-1-null mutant worms [3]. In fact, as shown in Figure 5, RNAi directed against genes rad-50, cand-1, cct-1, fat-2, and akt-1, encoding candidate PIP3-binding proteins, extended the length of time that adult worms could survive a lethal oxidative stress (5-mM H2O2), emulating the greatly enhanced peroxide-resistance of age-1(mg44) F2 adults [3]. Moreover, RNAi targeting rad-50, cand-1, fat-2, and dhc-1 rescued 92-100% of the paralysis that otherwise progressively afflicted worms expressing Aβ1-42 in body-wall muscle (Figure 6). RNAi directed against akt-1, although also significant, was less effective (69% rescue) ― perhaps due to functional redundancy between AKT-1 and AKT-2 proteins. Two of these RNAi treatments were previously reported to increase longevity, and three (targeting akt-1, cand-1 and rad-50) produced moderate increases in lifespan (9-20%) in our hands.
In view of the importance of PI3KI as a driver of cell proliferation, inhibitors have been actively sought as potential chemotherapeutic agents for cancer. In pursuit of novel anti-cancer drugs, and equally in seeking drugs to prevent or ameliorate Alzheimer’s and other age-dependent diseases, potential therapeutic benefits of PI3KI inhibitors have been overshadowed by the virtual certainty that they would also be detrimental to stem cell niches. This concern has motivated our search for PIP3-binding proteins as alternative targets downstream of PI3KI. Among these, AKT is believed to drive most of the proliferative effects of PI3K [45], whereas RAD50, CAND1, FAT2 and TCT1 constitute novel targets that may preserve survival benefits of PI3KI disruption, uncoupled from blockage of cell proliferation.
Materials and Methods
C. elegans strains and culture
Wild-type Bristol N2 [DRM stock], DR26 [daf-16(m26)], AM141 (unc-54p/Q40:: YFP), CL4176 (smg-1ts [myo-3/Aβ1-42/long 3'-untranslated region (UTR)]), and NL5901 (unc-54p/α-synuclein::YFP), were obtained from the Caenorhabditis Genetics Center (CGC). Strain SR808 was created by outcrossing age-1(mg44) 6x into the N2-DRM background; strain DR26 was similarly outcrossed to create strain SR814 [3]. All strains were maintained at 20°C on 2% (w/v) agar plates containing nematode growth medium (NGM), overlaid with E. coli strain OP50. If induction is indicated, strain CL4176 was upshifted to 25°C at the L3-L4 transition to induce expression of the human Aβ1-42 transgene. To generate synchronized worms, well-fed C. elegans were lysed at day 3 post-hatch (adult day 1) to release unlaid eggs, which were plated on 100-mm Petri dishes containing NGM-agar seeded in a central area with E. coli (OP50) as described [3].
RNA interference
Targeted genes were subjected to RNAi knock-down by feeding worms (either from the time of hatching, or from the L4 (last larval) stage to avoid developmental effects of RNAi) on HT115 bacterial sublines from the Ahringer RNAi library [56]. Briefly, synchronized eggs were recovered after alkaline hypochlorite lysis and transferred to plates seeded with HT115 (DE3) bacteria, deficient in RNAse III and containing (a.) IPTG-inducible T7 RNA polymerase, and either (b.) the L4440 plasmid with a multiple cloning site (MCS) between two inward-directed T7 RNA polymerase promotors for “feeding vector (FV) controls”, or (c.) L4440 containing an exonic segment of the targeted gene, cloned into its MCS [56].
Isolation of membrane proteins
Synchronized day-3 C. elegans adults were collected after washing in S buffer, drained of excess liquid, and flash frozen in liquid nitrogen. The worm pellets were pulverized with a dry-ice-cooled mortar and pestle, and suspended in buffer with nonionic detergent (20-mM Hepes pH 7.4, 300-mM NaCl, 2-mM MgCl2, 1% NP40), and protease/phosphatase inhibitors (MilliporeSigma, Darmstadt, Germany) at 0°C. Worm or cell debris was removed by brief centrifugation of lysate (5 min. at 3000 rpm). Native membrane-associated proteins were isolated from lysates with ProteoExtract membrane purification kit (MilliporeSigma) following the manufacturer’s protocol, and either (a.) used for PIP3 binding (see next section), or (b.) suspended in Laemmli buffer containing 2% SDS (w/v) and 0.3-M β-mercaptoethanol, heated 5 min at 95°C to dissolve proteins, and electrophoresed on 4‒20% polyacrylamide gels (SDS-PAGE). Gels were stained with SYPRO Ruby (ThermoFisher) to visualize total protein.
Isolation of PIP3-binding membrane proteins
Isolated membrane proteins were pre-adsorbed to uncoated control beads; the unbound fraction was collected and incubated 6 h at 4°C with PIP2- or PIP3-coated agarose beads (echelon, Salt Lake City, UT). After extensive washing, bound proteins were eluted from PIP2- and PIP3-coated beads, suspended in Laemmli buffer containing 2% SDS (w/v) and 0.3-M β-mercaptoethanol, and heated 5 min at 95°C to dissolve proteins prior to separation on 4‒20% polyacrylamide/SDS gels as above.
Identification of membrane and/or PIP3-binding proteins
Proteins isolated from membranes, or membranes followed by PIP3-coated beads, were dissolved in Laemmli buffer as described above, and separated in one dimension on 1% SDS, 4‒12% acrylamide gradient gels. They were then stained with SYPRO Ruby (ThermoFisher) or Coomassie Blue to visualize total protein, and 1-mm slices were excised. Proteins were digested in situ with trypsin, and peptides analyzed by high-resolution LC-MS/MS with a Thermo-Velos Orbitrap mass spectrometer (ThermoFisher) coupled to a nanoACQUITY liquid chromatography system (Waters, Milford MA) as previously reported [20]. Proteins were identified by MASCOT (www.matrixscience.com) matching of peptide fragmentation patterns to a database of previously observed fragment patterns [20].
PIP3 feeding to “rescue” age-1-null worms
Very long-lived age-1(mg44) mutant worms are maintained as genetically mixed cultures, with a recessive visible-trait marker (dumpy) on a balancer chromosome carrying wild-type age-1. Synchronized worms that are not dpy/dpy (and thus not age-1+/+) are placed singly on individual nutrient-agar plates and classified based on the developmental rate of their progeny: heterozygotes (mg44/+) produce a majority of offspring that develop normally, reaching adulthood in 2.5 days, while all progeny of first-generation mg44/mg44 homozygotes are second-generation “F2” age-1(mg44) homozygotes that uniformly develop quite slowly (>8 days at 20oC, from hatch to the L4/adult moult [3]). These F2 mutants were fed 30-μM phosphatidylinositol 3,4,5-triphosphate di-C4 (echelon), beginning at 3 days post-hatch (as soon as they could be distinguished with certainty from their less long-lived siblings). PIP3 di-C4 differs from normal PIP3 by having very short (4-carbon) fatty-acid chains to improve water solubility.
Paralysis assays
CL4176 worms were synchronized as described above, and eggs were transferred to 60-mm NGM-agar plates seeded with either FV-control bacteria or RNAi-expressing bacteria to target each gene that encodes a protein of interest. Paralysis of worms with muscle expression of Aβ1-42 was assayed as described previously [36]. Briefly, worms were upshifted from 20 to 25°C at the L3-L4 transition, and triplicate groups of 50‒100 worms were scored 29 h later. Alternatively, age-dependent paralysis was monitored over 10-13 days post-hatch, maintaining worms at 20°C without upshift. Paralysis was defined by movement of the head, but not the body, in response to a touch stimulus.
Aggregation assays for Q40::YFP and α-synuclein::YFP
AM141 worms with muscle expression of an unc-54p/Q40::YFP transgene [27] were synchronized and fed from hatch with RNAi targeting each individual gene that encodes a PIP3-binding protein, or empty feeding vector (FV controls). They were assessed by imaging yellow fluorescence, 4 days post-hatch as described [36, 56]. NL5901 worms, with muscle expression of an unc-54p/α-synuclein::YFP fusion protein [57], were grown on RNAi (or FV) bacteria as described above and imaged at days 9 and 10 post-hatch. To record images, worms were immobilized on glass slides in S buffer containing 0.3% (w/v) sodium azide to block muscle contraction, and fluorescence images were captured on a DP71 camera mounted on a BX51 fluorescence microscope (Olympus, Tokyo) with a 10x objective. YFP-containing aggregates (fluorescent foci) were counted for >15 worms (5-10 fields) per group with dotcount (reuter.mit.edu/software/ dotcount). Statistical significance of differences in counts/worm were based on 5-10 fields per group, by 2-tailed heteroscedastic t tests.
Longevity survivals
Synchronized eggs (or L4 larvae, to avoid developmental effects) were plated on control bacteria containing an empty feeding-vector plasmid, or on RNAi bacteria selected from the Ahringer library [56] transcribing double-stranded RNA of an exonic segment from a PIP3-binding protein. Worms were transferred to fresh plates daily for 7 days, and on alternate days thereafter, scoring worms as alive if they moved spontaneously or in response to gentle prodding [3]. Worms lost for reasons other than natural death were censored (removed from mortality calculations) from the date of first annotation onward.
Hydrogen peroxide stress survivals
Worms were hatched and maintained on RNAi or control plates as in the preceding section. Day-1 adult worms (50 worms, 24 h after the L4/adult molt) were washed free of bacteria and incubated at 20oC in 300 μl of 5-mM H2O2 in a 24-well plate. Worms were scored at hourly intervals for survival based on movement in response to touch [36] to assess each RNAi for effect on H2O2-stress survival.
Computer modeling of protein structures
Sequences of all proteins were compiled from WormBase and UniProt databases, and used to perform BLASTP searches to retrieve known structures from PDB (Protein Data Base). The 3-dimensional structures of candidate proteins were modeled in two ways. In the case of nematode proteins for which a reasonable template exists (i.e. ≥70% identity to a protein of known structure), template modeling was performed in MODELLER 9.13 (salilab.org/modeller) with default parameters [36]. Proteins lacking good templates were modeled in I-TASSER (zhanglab.ccmb.med.umich.edu/I-TASSER) to predict structures by ab initio “multi-threading” methods [58]. Ramachandran plots were generated for all structures generated in Modeller or I-TASSER using the RAMPAGE server (mordred.bioc.cam.ac.uk/~rapper/rampage.php), and amino acids in dis-allowed regions were loop-refined using MODELLER. This process was continued iteratively until the entire plots fell within permitted limits. The final minimum-energy conformers were then used for further docking analyses as described.
Protein docking to PIP3 and PIP2
The structure of PIP3 (Pubchem database) was processed by truncation of fatty-acyl chains using ChemSketch, and hydrogen atoms added as required with MGL Tools and Python Molecular Viewer while monitoring torsions. The PIP2 structure was then generated from that of PIP3 by removing the 3-phosphate. AutoDock Vina 4.2 (http://vina.scripps.edu) was used with default parameters [59] to predict interactions of candidate proteins with PIP3 and PIP2, and to score the complexes for interaction energies. For control interactions, protein structures were retrieved at random from the PDB database (http://www.rcsb.org). Heteroatoms (ligand), if present in downloaded structures, were manually removed before docking, as above, in AutoDock Vina 4.2 (run with Raccoon interface on a 32-core Linux cluster). To avoid bias, entire proteins were made available for docking. Grid dimensions were defined manually for each interaction. The Gibbs free energy of binding was calculated as ΔGbinding = ΔGvdW + ΔGelec + ΔGH-bond + ΔGdesolv + Δgtors, where ΔGvdW = the Lennard-Jones van der Waals potential with 0.5Å smoothing; ΔGelec = the Solmajer-Mehler distance-dependent dielectric potential; ΔGH-bond = hydrogen-bonding potential with Goodford directionality; ΔGsolv = charge-dependent version of Stouten pairwise atomic solvation energy; and Δgtors is a function of the number of rotatable bonds in the ligand only (see http://autodock.scripps.edu for details).
Statistical analyses
Differences between groups were assessed for significance by the Fisher-Behrens heteroscedastic t test (appropriate to samples of unequal or unknown variance). Differences in relative peptide abundance, based on spectral counts relative to the total per sample, were assessed for significance by chi-squared or Fisher exact tests. Significance of longevity-survival differences was ascertained by Gehan-Wilcoxon log-rank tests. Significance of differences in peroxide-stress survival was assessed by Fisher exact tests at the earliest assay time for which control survival was < 15%.
acknowledgments
The authors thank the Proteomics Core facility of the University of Arkansas for Medical Sciences, Little Rock AR, for protein analyses; and Bharat Raghavan (Shresid Inc., Little Rock AR) for generous help in construction of high-performance computing servers.
Conflicts of Interest
The authors have no competing interests.
Grant support
The work reported here was supported by grants to RJSR from the Life Extension Foundation and the U.S. Veterans Administration (Merit Review and Senior Research Career Scientist Award); and to SA from NIH/NIA, a subaward of grant P30 AG028718 (J. Wei, P.I.).
REFERENCES
1. Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988; 118: 75-86.
2. Bharill P, Ayyadevara S, Alla R, Shmookler Reis RJ. Extreme depletion of PIP3 accompanies the increased life span and stress tolerance of PI3K-null C. elegans mutants. Front Genet. 2013; 4: 34.
3. Ayyadevara S, Alla R, Thaden JJ, Shmookler Reis RJ. Remarkable longevity and stress resistance of nematode PI3K-null mutants. Aging Cell. 2008; 7: 13-22.
4. Kriplani N, Hermida MA, Brown ER, Leslie NR. Class I PI 3-kinases: Function and evolution. Adv Biol Regul. 2015; 59: 53-64.
5. Park WS, Heo WD, Whalen JH, O’Rourke NA, Bryan HM, Meyer T, Teruel MN. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell. 2008; 30: 381-392.
6. Viaud J, Mansour R, Antkowiak A, Mujalli A, Valet C, Chicanne G, Xuereb JM, Terrisse AD, Severin S, Gratacap MP, Gaits-Iacovoni F, Payrastre B. Phosphoinositides: Important lipids in the coordination of cell dynamics. Biochimie. 2015; 125: 250-258.
7. Odorizzi G, Babst M, Emr SD. Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem Sci. 2000; 25: 229-235.
8. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2009; 9: 162-176.
9. Balla T, Szentpetery Z, Kim YJ. Phosphoinositide signaling: new tools and insights. Physiology (Bethesda). 2009; 24: 231-244.
10. Ortega-Molina A, Lopez-Guadamillas E, Mattison JA, Mitchell SJ, Muñoz-Martin M, Iglesias G, Gutierrez VM, Vaughan KL, Szarowicz MD, González-García I, López M, Cebrián D, Martinez S, et al. Pharmacological inhibition of PI3K reduces adiposity and metabolic syndrome in obese mice and rhesus monkeys. Cell Metab. 2015; 21: 558-570.
11. Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000; 100: 603-606.
12. Wenk MR, Lucast L, Di Paulo G, Romanelli AJ, Suchy SF, Nussbaum RL, Cline GW, Shulman GI, McMurray W, De Camilli P. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat Biotechnol. 2003; 21: 813-817.
13. Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006; 34: 647-662.
14. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008; 27: 5497-5510.
15. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011; 11: 289-301.
16. Popovics P, Gray A, Arastoo M, Finelli DK, Tan AJ, Stewart AJ. Phospholipase C-eta2 is required for retinoic acid-stimulated neurite growth. J Neurochem. 2013; 124: 632-644.
17. Ivey RA, Sajan MP, Farese RV. Requirements for pseudosubstrate arginine residues during autoinhibition and phosphatidylinositol 3,4,5-(PO(4))(3)-dependent activation of atypical PKC. J Biol Chem. 2014; 289: 25021-25030.
18. Lai CL, Srivastava A, Pilling C, Chase AR, Falke JJ, Voth GA. Molecular mechanism of membrane binding of the GRP1 PH domain. J Mol Biol. 2013; 425: 3073-3090.
19. Hart R, Stanley P, Chakravarty P, Hogg N. The kindlin 3 pleckstrin homology domain has an essential role in lymphocyte function-associated antigen 1 (LFA-1) integrin-mediated B cell adhesion and migration. J Biol Chem. 2013; 288: 14852-14862.
20. Boyken SE, Fulton DB, Andreotti AH. Rescue of the aggregation prone Itk Pleckstrin Homology domain by two mutations derived from the related kinases, Btk and Tec. Protein Sci. 2012; 21: 1288-1297.
21. Swanson KD, Tang Y, Ceccarelli DF, Poy F, Sliwa JP, Neel BG, Eck MJ. The Skap-hom dimerization and PH domains comprise a 3’-phosphoinositide-gated molecular switch. Mol Cell. 2008; 32: 564-575.
22. Campa F, Yoon HY, Ha VL, Szentpetery Z, Balla T, Randazzo PA. A PH domain in the Arf GTPase-activating protein (GAP) ARAP1 binds phosphatidylinositol 3,4,5-trisphosphate and regulates Arf GAP activity independently of recruitment to the plasma membranes. J Biol Chem. 2009; 284: 28069-28083.
23. Mas C, Norwood SJ, Bugarcic A, Kinna G, Leneva N, Kovtun O, Ghai R, Ona Yanez LE, Davis JL, Teasdale RD, Collins BM. Structural basis for different phosphoinositide specificities of the PX domains of sorting nexins regulating G-protein signaling. J Biol Chem. 2014; 289: 28554-28568.
24. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol. 2013; 31: 675-704.
25. Bernier LP, Blais D, Boue-Grabot E, Seguela P. A dual polybasic motif determines phosphoinositide binding and regulation in the P2X channel family. PLoS One. 2012; 7: e40595.
26. Hasegawa J, Tokuda E, Tenno T, Tsujita K, Sawai H, Hiroaki H, Takenawa T, Itoh T. SH3YL1 regulates dorsal ruffle formation by a novel phosphoinositide-binding domain. J Cell Biol. 2011; 193: 901-916.
27. Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002; 99: 10417-10422.
28. Deak F, Sonntag WE. Aging, synaptic dysfunction, and insulin-like growth factor (IGF)-1. J Gerontol A Biol Sci Med Sci. 2012; 67: 611-625.
29. Saatman KE, Contreras PC, Smith DH, Raghupathi R, McDermott KL, Fernandez SC, Sanderson KL, Voddi M, McIntosh TK. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp Neurol. 1997; 147: 418-427.
30. Perluigi M, Pupo G, Tramutola A, Cini C, Coccia R, Barone E, Head E, Butterfield DA, Di Domenico F. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim Biophys Acta. 2014; 1842: 1144-1153.
31. Gasparini L, Xu H. Potential roles of insulin and IGF-1 in Alzheimer’s disease. Trends Neurosci. 2003; 26: 404-406.
32. Zemva J, Schubert M. The role of neuronal insulin/insulin-like growth factor-1 signaling for the pathogenesis of Alzheimer’s disease: possible therapeutic implications. CNS Neurol Disord Drug Targets. 2014; 13: 322-337.
33. Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat Med. 2010; 16: 438-445.
34. Wang J, Richards DA. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol Open. 2012; 1: 857-862.
35. Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le BN, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003; 421: 231-237.
36. Ayyadevara S, Balasubramaniam M, Gao Y, Yu LR, Alla R, Shmookler Reis RJ. Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome-blocking complexes. Aging Cell. 2015; 14: 35-48.
37. Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003; 24: 415-420.
38. Nojima A, Yamashita M, Yoshida Y, Shimizu I, Ichimiya H, Kamimura N, Kobayashi Y, Ohta S, Ishii N, Minamino T. Haploinsufficiency of akt1 prolongs the lifespan of mice. PLoS One. 2013; 8: e69178.
39. Yuan Y, Kadiyala CS, Ching TT, Hakimi P, Saha S, Xu H, Yuan C, Mullangi V, Wang L, Fivenson E, Hanson RW, Ewing R, Hsu AL, et al. Enhanced energy metabolism contributes to the extended life span of calorie-restricted Caenorhabditis elegans. J Biol Chem. 2012; 287: 31414-31426.
40. Shmookler Reis RJ, Xu L, Lee H, Chae M, Thaden JJ, Bharill P, Tazearslan C, Siegel E, Alla R, Zimniak P, Ayyadevara S. Modulation of lipid biosynthesis contributes to stress resistance and longevity of C. elegans mutants. Aging (Albany NY). 2011; 3: 125-147. doi: 10.18632/aging.100275
41. Branicky R, Desjardins D, Liu JL, Hekimi S. Lipid transport and signaling in Caenorhabditis elegans. Dev Dyn. 2010; 239: 1365-1377.
42. Malmstrom RD, Watowich SJ. Using free energy of binding calculations to improve the accuracy of virtual screening predictions. J Chem Inf Model. 2011; 51: 1648-1655.
43. Thomas CC, Deak M, Alessi DR, van Aalten DM. High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol. 2002; 12: 1256-1262.
44. Wang Y, Oh SW, Deplancke B, Luo J, Walhout AJ, Tissenbaum HA. C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF-16/FOXO. Mech Ageing Dev. 2006; 127: 741-747.
45. Gami MS, Iser WB, Hanselman KB, Wolkow CA. Activated AKT/PKB signaling in C. elegans uncouples temporally distinct outputs of DAF-2/insulin-like signaling. BMC Dev Biol. 2006; 6: 45.
46. Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS ONE. 2010; 5: e8758.
47. Ayyadevara S, Tazearslan C, Alla R, Bharill P, Siegel ER, Shmookler Reis RJ. C. elegans PI3K mutants reveal novel genes underlying exceptional stress resistance and lifespan. Aging Cell. 2009; 8: 706-725.
48. Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007; 3: e56.
49. Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, Mackay VL, An EH, Strong R, Ladiges WC, Rabinovitch PS, Kaeberlein M, Kennedy BK. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012; 4: 144ra103.
50. Morimoto RI, Cuervo AM. Proteostasis and the aging proteome in health and disease. J Gerontol A Biol Sci Med Sci. 2014; 69 Suppl 1: S33-S38.
51. Nollen EA, Garcia SM, van HG, Kim S, Chavez A, Morimoto RI, Plasterk RH. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2004; 101: 6403-6408.
52. Bandyopadhyay B, Li G, Yin H, Kuret J. Tau aggregation and toxicity in a cell culture model of tauopathy. J Biol Chem. 2007; 282: 16454-16464.
53. Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, Rutkowski DT, Kaufman RJ, Ruse CI, Yates JR, III, Perrin S, Feany MB, Horwich AL. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci U S A. 2009; 106: 1392-1397.
54. Li D, Shi JJ, Mao CJ, Liu S, Wang JD, Chen J, Wang F, Yang YP, Hu WD, Hu LF, Liu CF. Alteration of dynein function affects alpha-synuclein degradation via the autophagosome-lysosome pathway. Int J Mol Sci. 2013; 14: 24242-24254.
55. Vidugiriene J, Vainauskas S, Johnson AE, Menon AK. Endoplasmic reticulum proteins involved in glycosylphosphatidylinositol-anchor attachment: photocrosslinking studies in a cell-free system. Eur J Biochem. 2001; 268: 2290-2300.
56. Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003; 30: 313-321.
57. van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008; 4: e1000027.
58. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010; 5: 725-738.
59. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010; 31: 455-461.