
INTRODUCTION
In multicellular organisms, the balance between cell death, proliferation and differentiation is crucial for the maintenance of organ development, tissue homeostasis, and aging. For many years, the three basic types of cell death, type I, II, and III, could be distinguished according to morphological, enzymological, and functional criteria. Type I, apoptotic cell death, is defined by an ensemble of morphological features including chromatin condensation, nuclear fragmentation, cell shrinkage, plasma membrane blebbing, and the formation of an apoptotic bodies. Type II, autophagic cell death, is a process by which cells generate energy and metabolites by digesting organelles or macromolecules. Type III, necrosis, is characterized by a lack of stereotypical morphological changes, but does eventually result in the rounding of the cell, cytoplasmic swelling, and the rupture of the plasma membrane and the spilling of the intracellular contents [1-2].
The field of cell death research has rapidly developed, resulting in the recommendation of updated cell death classification criteria by the Nomenclature Committee on Cell Death [2]. For example, necrosis has for a long time been considered an accidental mode of cell death. However, it was recently recognized that a form of cell death morphologically classified as necrosis could also be regulated in a programmed manner via defined signal transduction pathways (termed necroptosis) [3-5]. Multiple lines of evidence indicate that necrosis can be a programmed event: (I) cell death with a necrotic appearance can contribute to embryonic development and adult tissue homeostasis; (II) necrotic cell death can be induced by ligands that bind to specific plasma membrane receptors, (III) necrosis can be regulated by genetic, epigenetic, and pharmacological factors [6]; (IV) the cellular disintegration phase of necrosis is characterized by an identical sequence of sub-cellular events, including oxidative burst, mitochondrial membrane hyperpolarization, lysosomal membrane permeabilization and plasma membrane permeabilization, although with different kinetics [7]; and (V) the inactivation of caspases causes a shift from apoptosis either to cell death morphologies with mixed necrotic and apoptotic features or to full-blown necrosis [8].
The molecular mechanisms involved in necroptosis have been intensively studied in recent years. In principle, a multitude of different stimuli can initiate necroptosis, comprising mainly of three phases of signal transduction, including an initiation and an execution phase associated with the loss of cell and organelle integrity. The execution necroptosis phase involves activation of specific death mediators, such as receptor-interacting protein kinases (RIPKs) and mixed-lineage kinase domain-like protein (MLKL) [9-10].
Accumulating evidence indicates that necroptosis is involved in the regulation of cancer [11-16]. It is widely accepted that evasion of cell death is one of the hallmarks of cancer [17-18]. Many lines of clinical and experimental evidence have demonstrated that defects in cancer cell death are the most frequent causes of therapeutic resistance, and thus exploring cancer cell death might inform development of strategies to overcome therapeutic resistance. Although the molecular mechanisms underlying necroptosis need to be further elucidated, it is becoming clear that further insights into the signaling networks involved in regulation of necroptosis will likely have important implications for the exploitation of this form of regulated cell death for the diagnosis or treatment of cancer in the complex tumor microenvironment. With these aims in mind, in this review, we summarize the role of necroptosis in tumorigenesis, activation of anti-tumor immunity, and cancer therapy.
Mechanisms and regulation of necroptosis
Considering the emerging significance of necroptosis in cancer, a better understanding of the molecular mechanisms underlying necroptotic signaling will likely have important implications for the development of novel methods to interfere with necroptosis in cancer. In principle, a multitude of different stimuli can initiate necroptotic cell death, which mainly comprises three phases of signal transduction, including an initiation and an execution phase, finally causing the loss of cell and organelle integrity and cell death (Figure 1).
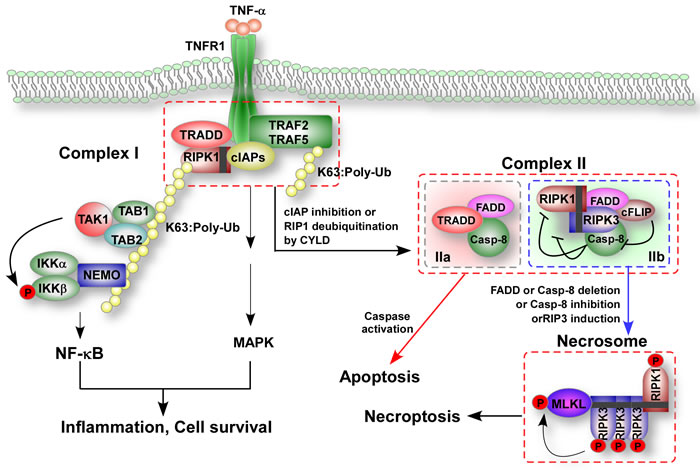
Figure 1: TNF-induced formation of apoptotic and necroptotic signaling complexes. After ligand binds to the receptor, the intracellular tails of tumor necrosis factor receptor 1 (TNFR1) recruit multiple proteins to form the membrane-proximal supramolecular structure complex I including TNFR1 associated death domain protein (TRADD), receptor-interacting protein kinase-1 (RIPK1), cellular inhibitors of apoptosis (cIAPs), the E3 ubiquitin ligases TNF-receptor-associated factor 2 and 5 (TRAF2 and TRAF5). Lys63-linked polyubiquitination (K63-poly Ub) of RIPK1 in complex I mediated by cIAP ligases is crucial for the recruitment of nuclear factor-κB (NF-κB) essential modulator (NEMO), a regulatory subunit of IκB kinase (IKK) complex that in turn activates NF-κB and mitogen-activated protein kinases (MAPKs). Deubiquitination RIPK1 by cylindromatosis (CYLD) or inhibition of cIAP proteins promote the conversion of complex I to complex II and inhibits NF-κB activation. Complex II contains RIPK1, Fas-associated protein with death domain (FADD), caspase-8, cellular FLICE-inhibitory protein-L (cFLIPL), RIPK3 and TRADD. Caspase-8 becomes activated in complex II and initiates apoptosis, whereas cFLIPL can prevent activation of caspase-8. In cells with high levels of RIPK3 expression, RIPK3 enters complex II via interaction with RIPK1 after stimulation. The RIPK3-containning complex is called complex IIb or the necrosome. In the presence of cFLIPL, caspase-8 is unable to initiate apoptosis but can cleave RIPK1 and RIPK3 and thus inhibits necroptosis. Depletion of FADD or caspase-8, inhibition of caspase-8 or induction of RIPK3 can free RIPK1-RIPK3 from inhibition and initiate necroptosis by mixed-lineage kinase domain-like protein (MLKL) of TNF-treated cells.
Initiation of necroptosis
Necroptosis can be elicited by a range of stimuli, from cytokines, viral infection, chemicals, or damage-associated molecular patterns (DAMPs), to several forms of physicochemical cellular stress [19]. Different necroptotic stimuli are recognized or sensed by specific receptors or sensors on the cell surface or cell interior. A range of receptor-sensor complexes can initiate the necroptotic response to different stimuli, although the nature of some of these complexes is presently unknown. We focus on the components of TNF-α signaling, because this signaling pathway is the most extensively studied inducer of necroptosis [9]. Under some conditions, TNF signaling occurs primarily through TNF receptor 1 (TNFR1), a potent inducer of induced cell death. However, early evidence demonstrated that TNF induces caspase-independent cell death by a mechanism involving activation of RIPK1 [3, 20-21]. Also important is the identification of necroptotic inhibitor necrostatin-1 (Nec-1) targeting RIPK1 that indicated that TNF-α induced necroptosis is a kinase-regulated process [16, 22]. In addition, RIPK3 was reported to be an essential regulator of TNF-α induced necroptosis [22-24], and TNF-α stimulation was reported to induce the formation of a necrosome in which RIPK3 is activated to interact with RIPK1 via the RIP homotypic interaction motif (RHIM) initiating necroptosis [23-25].
Besides TNF-α, there are five different stimulators of necroptosis. (I) Fas or tumor necrosis factor related apoptosis inducing ligand receptor (TRAILR): stimulation of Fas or TRAILR induces formation of the receptor-bound death inducing signaling complex that triggers caspase-8 mediated apoptosis independent of RIPK1. Under particular conditions, such as the absence of cellular inhibitors of apoptosis (cIAPs), which favor the recruitment of RIPK1 to Fas [26], and the formation of a cytosolic ripoptosome complex [27], which mediates necroptosis when caspase-8 is blocked. (II) Toll-like receptor-4 (TLR4) and Toll-like receptor-3 (TLR3): TLR4 or TLR3 stimulation triggers formation of the necrosome through the RHIM containing adapter Toll/IL-1 receptor domain containing adaptor protein inducing interferon (IFN)-β (TRIF), resulting in RIPK3 dependent necroptosis in which the role of RIPK1 depends on the cellular context. (III) double-stranded DNA viruses such as murine cytomegalovirus (MCMV): DNA dependent activator of IFN regulatory factors recognize viral double-stranded DNA and, through its RHIM domain, recruits RIPK3 to induce formation of the necrosome without RIPK1, triggering RIPK1 independent RIPK3 kinase activity dependent necroptosis. (IV) IFN-α and IFN-β: IFN-α and IFN-β induce necroptosis through their cognate receptors, interferon alpha receptors (IFNRs), leading to activation of the Janus kinase/Signal transducer and activator of transcription (JAK/STAT). In cells that were deficient in nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling, IFN-γ promoted accumulation of mitochondrial reactive oxygen species (ROS) and eventual loss of mitochondrial membrane potential that ultimately leads to necroptosis; and (V) polyinosine-polycytidylic acid (Poly (I:C)): Poly (I:C)-TLR3 stimulation in the absence of zVAD-fmk induced TRIF mediated necroptosis (Figure 2).
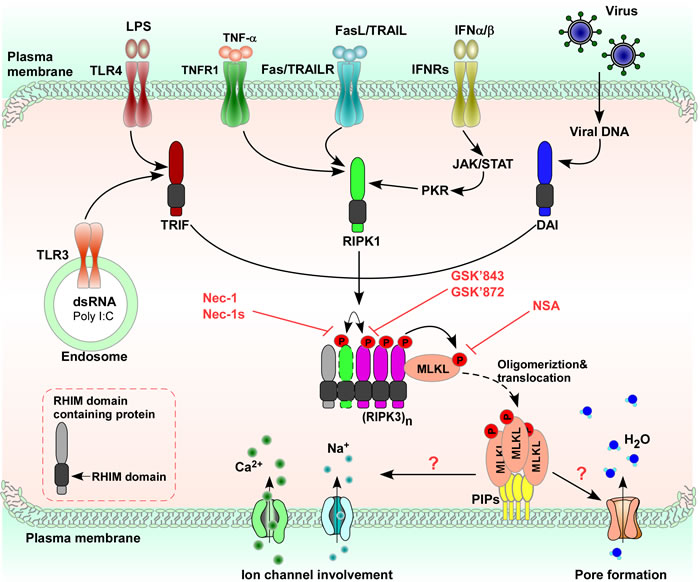
Figure 2: Necroptosis is induced by various stimuli. Necroptotic stimuli such as TNF-α, FasL/TRAIL, IFN-α/IFN-β, pathogen-associated molecular patterns (LPS, poly (I:C)) via TLR activation or virus-mediated activation of DAI are recognized or sensed by specific receptors or sensors on the surface of or inside cells, and mediate the initiation of the necroptotic response to different stimuli, although the nature of some of these complexes is currently unknown. Formation of the necroptotic-signaling complex determines cell date. Different necroptotic complexes are found after ligation of different receptors or under genotoxic stress. When different necroptotic stimuli induced necroptosis, a complex called the necrosome is formed leading to RIPK3 oligomerization. MLKL could then modify ion influx, changing osmotic pressure, and/or forming a pore, thus causing plasma membrane rupture. In addition, necroptosis can be pharmacologically inhibited by Nec-1 (RIPK1 inhibitor), GSK- 843/872/840 (RIPK3 inhibitors), and NSA (MLKL inhibitor).
Execution of necroptosis
Efforts to further explore the execution of necroptosis include establishment of a robust cell-based model of necroptosis. By treating human colon cancer HT-29 cells with TNF-α, about 200,000 chemical compounds were screened for their ability to inhibit necroptosis, and MLKL was identified as a critical substrate of RIPK3 during the induction of necroptosis [28]. Meanwhile, the fact that MLKL pseudokinase is a substrate of RIPK3 required for necroptosis sheds light on the mechanisms involved in executing necroptotic cell death downstream of RIPK3 [29], as RHIM-dependent oligomerization and intramolecular autophosphorylation of RIPK3 results in the recruitment and phosphorylation of MLKL at Ser 345, Ser 347, and Thr349 [30-32]. Further studies demonstrated that the phosphorylation of MLKL at Ser 345 is not required for interaction between RIPK3 and MLKL in the necrosome, but is essential for MLKL translocation, accumulation in the plasma membrane, and consequent necroptosis [33]. Taken together, two non-exclusive models are proposed for the executioner mechanism of MLKL: one as a platform at the plasma membrane for the recruitment of Ca2+ or Na+ ion channels [34-35], and the other as a direct pore-forming complex that is recruited through binding of the amino-terminus of the 4-helical bundle domain of MLKL to negatively charged phosphatidylinositophosphates [36-38].
In addition, phosphoglycerate mutase 5 (PGAM5), a mitochondrial phosphoglyceratemutase, was identified to be another necrosome-associated protein that can regulate dynamic-related protein (Drp1) through its dephosphorylation [39-40]. It is noteworthy that the two variants of PGAM5 (PGAM5S and PGAM5L) have different functions during necroptosis, even though they are both required for necroptosis execution. After the necrosome core is formed, PGAM5L binds to the necrosome, unaffected by the MLKL inhibitor necrosulfonamide (NSA); however, NSA blocks PGAM5S. In addition, multiple leading cell death laboratories have recently reported that PGAM5 is dispensable for necroptosis [41-42]. Therefore, although many cellular events have been reported to act downstream of the necroptotic-signaling complex to execute necroptosis, the role of PGAM5 during necroptosis remains largely elusive and somewhat controversial. Further investigation will be required to clarify the importance of these executors of necroptosis.
Mechanisms of necroptosis
After TNF-α binds to the receptor TNFR1, the intracellular tails of TNFR1 recruit multiple proteins to form the membrane associated protein Complex I, which contains TNFR1 associated death domain protein (TRADD), RIPK1 and the E3 ubiquitin ligases TNF-receptor-associated factor 2 and 5 (TRAF2 and TRAF5), cIAPs, and the linear ubiquitin chain assembly complex (LUBAC) [3, 43-44]. This complex provides a platform for the recruitment of downstream kinases and effector proteins to activate NF-κB and mitogen-activated protein kinase (MAPK) [45-51]. NF-κB and MAPK are believed to initiate a survival pathway because they induce expression of some genes encoding cytoprotective molecules [52].
Under some conditions, the intracellular tails of TNFR1 also induce apoptosis through cytosolic complex IIA (TRADD, Fas-associated protein with death domain (FADD), and caspase-8) and IIB (RIPK1, RIPK3, FADD, caspase-8, and cellular FLICE-inhibitory protein-L (cFLIPL)), and in particular conditions or cells, necroptosis can be executed by necrosomes including RIPK1, RIPK3, and MLKL [53]. Destabilization of complex I leads to the formation of a second cytosolic complex IIA, which induced apoptosis and inhibits NF-κB activation [54-55]. In conditions such as TNF stimulation in the inhibition of cIAPs [52] or deubiquitination of RIPK1 by cylindromatosis (CYLD) [56-58], a cytosolic IIB complex forms, precipitating apoptosis. In cells with high levels of RIPK3 and MLKL expression, or under the conditions when caspase-8 activity is reduced, blocked or absent; complex IIB may form the necrosome [59].
Mechanisms determining the type of cell death
It is clear that programmed cell-death signaling pathways share some common components. For example, the same death stimuli can trigger different modes of cell death according to the molecular complexes present in the pathway. Different modes of cell death can also occur simultaneously or separately depending on the cellular circumstances.
Currently, the precise mechanisms that determine progression to necroptosis remain poorly understood. Numerous studies have suggested that expression of RIPK3 and MLKL correlates with sensitivity to necroptosis [24, 36, 60-64], however, a potential drawback of these studies is that RIPK3 and MLKL expression were compared between healthy and inflamed tissues. It is, therefore, difficult to conclude that RIPK3 and/or MLKL over-expression have primary causal functions.
Caspase-8 is the most crucial factor for preventing necroptosis. TNF-α induced signaling towards necroptosis is prevented by caspase-8, and it is reported to inhibit necroptosis by cleaving RIPK1 [65] and RIPK3 [66], as well as CYLD [67]. Currently, sensitization to necroptosis is achieved through a genetic defect compromising FADD-caspase-8 signaling and thus inhibiting apoptosis [68-71]; and inhibition of caspase-8 causes necroptosis [24], suggesting that whether a cell dies by apoptosis or necroptosis depends on caspase-8 activity.
In addition, ubiquitination or deubiquitination of RIPK1 can regulate the TNFR1 signaling pathway, driving necroptosis in vitro and in vivo [60, 68, 72]. In contrary, several brakes on RIPK1, affecting the presence or absence of RIPK1 and its post-translational modifications such as ubiquitylation and phosphorylation, can also inhibit necroptosis. Although ubiquitination has clearly emerged as a major regulatory mechanism, the substrates, exact linkage composition of the different chains, and the precise roles of the diverse related enzymes remain to be clarified [73-75].
Necroptosis in tumorigenesis
Necroptosis was recently reported to play a very important role in tumorigenesis as a backup cell death mechanism in cancer cells. Hitomi et al. [72] used a genome-wide screen using small interfering RNAs to delineate a cellular signaling network that regulated necroptosis and implicated two suppressor genes, CYLD and EDD1, and four Ras related proteins in regulation of necroptosis, which suggested a possible function of necroptosis in tumorigenesis.
Yang et al. [76] reported that the ratio of RIPK3-r, a truncated splice variant of RIPK3, to RIPK3 was significantly increased in colon and lung cancers relative to matched normal tissues, indicating that RIP3-r may be a major splice form associated with tumorigenesis. Furthermore, the RIPK3 gene is located on chromosome 14q11.2, a locus frequently altered in many cancers including nasopharyngeal carcinoma and T cell leukemia/lymphoma [77]. In non-Hodgkin lymphoma, polymorphisms in the RIPK3 gene were identified and found to correlate with increased risk of tumors [78].
Mutations in the CYLD gene in tumorigenic epidermal cells increase the aggressiveness of carcinomas, mainly by enhancing expression of angiogenic factors, thereby playing a key role in epidermal cancer malignancy [79-80]. In chronic lymphocytic leukemia cells (CLL), RIPK3 and CYLD were downregulated and lymphoid enhancer-binding factor 1 (LEF1) acts as a transcription repressor for CYLD [81]. Taken together, necroptosis may play a very important role in tumorigenesis [82].
Necroptosis backs up tumor immune activation
The idea that cell death may precede, trigger or amplify immunity has recently gained increasing attention. Similar to apoptotic cells, necroptotic tumor cells can induce anti-tumor immunity, necroptotic tumor cells can be cleared by innate immune phagocytic cells and engulfed by dendritic cells, macrophages, monocytes, and neutrophils, inducing release of pro-inflammatory cytokines and chemokines, upregulation of stimulatory molecules and enhanced cross-presentation, and eventual trigger of adaptive immune responses (Figure 3).
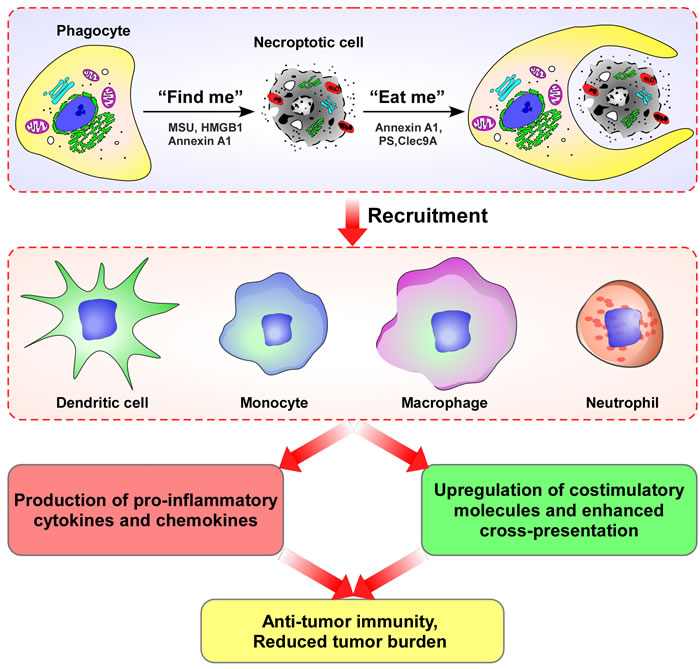
Figure 3: The concept that cell death may precede, trigger or amplify immunity has gained increasing attention. Immune activation phagocytic cells (dendritic cells, macrophages, monocytes, and neutrophils) can identify and engulf necroptotic tumor cells, inducing production of pro-inflammatory cytokines and chemokines, up-regulation of stimulatory molecules and enhanced cross-presentation; and, eventually, triggering of adaptive immune response.
Necroptotic cell clearance
Higher-level organisms have developed impressively efficient immune mechanisms to clear dying cells, for which the capability to distinguish dying from viable cells is crucial [83]. The sophisticated process of apoptotic cell removal involves several distinct phases, phagocyte recruitment, the engulfment of target cells, and the post-phagocytic response [84-86]. Phagocyte recruitment is accomplished by the release of soluble “find-me” signals from the dying cell [87-88]. Similar to apoptotic cells, the most likely candidates for necroptotic cell derived “find-me” signals are monosodium urate (MSU) crystals [89], high mobility group box 1 protein (HMGB1) [90], and annexin A1 [91-92]. When phagocytes reach the dying cells, they identify and recognize “eat-me” signals, which are exposed on the surface of the dying cell. Specific “eat-me” signals of necroptotic cells include annexin A1 [92-93], phosphatidylserine (PS) [94-95], and the C-type lectin Clec9A [96-98]. After dying cells are engulfed, phagocytic cells, including dendritic cells, macrophages, monocytes, and neutrophils can produce pro-inflammatory cytokines and chemokines, up-regulate stimulatory molecules and enhance cross-presentation, eventually triggering adaptive immune responses [99-100]. In addition, Martinez et al found that LC3-associated phagocytosis may play a key role in the clearance of dying cells and inflammation in the control of systemic lupus erythematosus [101]. Of course, further studies will be required to clarify the mechanisms of necroptotic cell clearance.
Effect of necroptosis on pro-inflammatory cytokine production
Necroptosis is thought to directly trigger inflammation through a massive release of DAMPs including HMGB1, interleukin family cytokines, adenosine triphosphate (ATP), and so on, which are released by necrotic cells [102]. However, necrotic cells can inhibit inflammatory reactions under some conditions [103-104]. For example, macrophages can engulf necroptotic L929 cells without producing inflammatory cytokines [98], highlighting an unexpected complexity in the immune response to necrotic cells. Theoretically, acute release of DAMPs could enhance the pro-inflammatory effect of necroptosis; however, the role of specific DAMPs in necroptosis induced in inflammation has not been functionally validated. While DAMPs appear to be involved in necroptosis-induced inflammation, their role awaits in vivo experimental validation.
Necroptotic cell induction of anti-tumor immune response
Dying cells induce DAMPs and initiate adaptive immunity by providing both antigens and inflammatory stimuli for DCs, which in turn activate CD8+ T cells through antigen cross-priming [105-106]. For example, IFN-α/β predominantly affects macrophages, DCs, and natural killer (NK) cells; inducing their activation and/or maturation, upregulating of chemokine expression, enhancing antigen-presentation and cross-presentation by DCs and a robustly augmenting induction of adaptive immune responses [107].
Anticancer agents induce a type of cell stress and death that is immunogenic; directly killing cancer cells, and using dead cells to provoke innate and adaptive immunity to attack other cancer cells [108]. For example, some studies demonstrated that stimulation of cervical cancer cells with PolyI:C, induced necroptotic cell death, then necroptotic cancer cells released IL-1α, which was required for the strong activation of DCs to produce IL-12, a cytokine critical for anti-tumor immune responses [109-110]. Kang et al. [111] implicated RIPK3-PGAM5-Drp1 signaling in NKT cell activation, and further suggested that RIPK3-PGAM5 signaling may mediate crosstalk between mitochondrial function and immune signaling. Aaes et al. reported that necroptotic cancer cells release DAMP and promote DC maturation, cross priming of cytotoxic T cells, and production of IFN-γ in response to tumor antigen stimulation [112]. Recently, using the model of cervical cancer, Smola demonstrated that PolyI:C driven immunogenicity strictly depends on the necroptosis regulator RIPK3 in neoplastic cells, suggesting RIPK3 as a novel predictive marker for personalization of cancer immunotherapy [113].
Although necrosis may induce effective immune responses, out of control necrosis could potentially cause irreversible tissue damage [114]. Additionally, the immune inflammatory cells recruited by the inflammatory factors released by necrotic cells may promote angiogenesis and cancer cell proliferation [115]. By tolerating some degree of necrotic cell death, invasive and metastatic tumors may gain growth-stimulating factors from the recruited tumor-promoting inflammatory cells [6]. Therefore, role of necroptosis of cancer and anti-tumor immune responses may be difficult to elucidate.
Necroptosis in T-cell expansion
Several reports indicate that necroptosis regulates the antigen-induced proliferation of T cells required for peripheral T cell homeostasis and T cell survival in response to activation stimuli [116]. For example, mice lacking caspase-8, or expressing a dominantly interfering form of FADD unable to recruit caspase-8 in T cells, exhibited impaired T cell homeostasis and diminished peripheral T cell numbers [117]. Impaired expansion of T cells after Fyn-T cell receptor (TCR) activation was observed in T cells deficient in caspase-8 [118-119] or FADD [120], as well as in FADDdd-expressing T cells [121-122]. Furthermore, blockade of RIPK1 through Nec-1 restores the expansion defect in caspase-8 deficient and FADDdd-expressing T cells [123-124], as does the genetic knockdown of RIPK1 in FADD deficient mice [72]. Similarly, loss of RIPK3 can rescue defective T cell proliferation of caspase-8-/- or FADDdd mice [125-126], indicating that necroptotic signaling in T cells is regulated by caspase-8.
Osborn et al. [120] demonstrated that necroptosis occurs during the late stage of normal T-cell proliferation, and that this process is greatly amplified in FADD-deficient T cells. Furthermore, in a thymidine kinase transfected tumor mouse model, the necrosis induced by a vascular target agent ZD6126, impaired the function of tumor-specific CD8+ T cells and abrogated a robust tumor specific immune response [127]. Cho et al. demonstrated that Nec-1 can inhibit RIPK1 dependent and independent necroptosis, and that a high dose of Nec-1 will inhibit other signal transduction pathways such as that for T cell receptor activation [128]. In addition, regulatory T cells (Tregs) are potent immune regulators and play a key role in maintaining peripheral tolerance. The contribution of two forms of programmed cell death, apoptosis and RIPK dependent necroptosis, to Treg-mediated suppression were evaluated, and it was found that Tregs can’t mediate suppress apoptosis or necroptosis via programmed cell death pathways [129]. Therefore, the capacity of necroptosis to trigger tumor immunity requires further investigation.
Necroptosis in cancer therapy
Necroptosis may be linked to not only tumorigenesis and anti-tumor immunity, but also to the success of cancer treatments. Characterization of the precise mechanisms underlying necroptosis and the molecular switches between apoptosis and necroptosis, may have major therapeutic implications [130-131]. In particular, we will focus on converging and diverging evidence implicating necroptosis in cancer therapies, and hope that research into necroptosis will provide new targets for effective therapies.
Necroptosis as a cancer therapeutic target
As discussed above, necroptosis is often impaired during tumorigenesis, but can be engaged by targeted pharmacological approaches. It is also clear that necroptosis plays a pivotal role in the pathogenesis of cancer. Therefore, characterization of the signal transduction pathways underlying necroptosis may highlight therapeutic targets.
Compounds and anticancer drugs
A growing list of compounds and anticancer drugs with various primary mechanisms of action, have been reported to initiate programmed necrosis or necroptosis in different cancer cells via mediated RIPK1, RIPK3, MLKL, and HMGB1 [132-166]. However, most studies on cancer and necroptosis are based on in vitro experiments, thus the efficacy and safety of these compounds and anticancer drugs still need investigation in vivo (Supplementary File 1).
Kinase inhibitors
Necroptosis has been implicated in the anti-tumor activity of several kinase inhibitors. Bl2536, a small molecule inhibitor of the mitotic kinase pololike kinase 1 (Plk1), has been reported to induce necroptosis in androgen-resistant prostate cancer cells [167]. Compound C, a widely used 5’-AMP-activated protein kinase inhibitor was described to induce glioma cell death through several mechanisms including necroptosis [168]. A cell permeant urokinase plasminogen activator system inhibitor 5’-benzylglycinyl-amiloride (UCD38B) also induces cell cycle independent and caspase-independent death of necroptotic glioma cells and breast cancer cells via apoptosis-inducing factors, independent of poly (ADP-ribose) polymerase and H2AX activation [169-170]. The formation of ATG5 deficient autophagosomes in response to sorafenib promotes interaction of p62 with RIPK, leading to cell death by necroptosis [171-172]. In addition, the combination of Givinostat and Sorafenib caused sustained production of ROS and activation of necroptotic relapsed/refractory Hodgkin’s lymphoma cell death [172].
Radiation therapy and chemotherapy
In addition to the drugs and kinase inhibitors that promote necroptosis, radiation and/or chemotherapy can also induce necroptosis of cell death [173]. In tumor cells of epithelial origin, radiotherapy induces limited apoptosis, but when combined with hyperthermia has been reported to stimulate necroptosis [174-175]. Furthermore, active caspase-8 induces apoptosis in response to low-doses radiation and inhibits necrosis by cleaving RIPK1. When activation of caspase-8 was reduced by high doses radiation, the RIPK1/RIPK3 necrosome II was formed. These complexes induce necroptosis through the caspase-3-independent pathway, mediated by calpain, cathepsin B/D, and apoptosis-inducing factor (AIF) [176]. MyD88 may determine whether UV irradiation causes apoptosis or necroptosis [177]. In addition, pan-caspase inhibitors can sensitize resistant colon cancer cells to 5-FU-induced necroptosis [178]. As radiation equipment and techniques, and chemotherapies improvement, further studies will be required to explore the potential for anticancer strategies to manipulate necroptosis with minimal side effects.
Others
MicroRNAs (miRNAs) are highly conserved, small noncoding RNA molecules that function to regulate a wide variety of cellular processes including cell proliferation and differentiation, fate determination, signal transduction, organ development, host-viral interactions, tumorigenesis and tumor progression [179-180]. While the mechanisms by which miRNA regulate necroptotic cell death are not well understood, miRNA-874 was reported to enhance necroptosis by targeting caspase-8, which acts as a key modulator of the transition between apoptosis and necroptosis [181]. However, how miRNAs regulate other key necroptotic factors, including RIPK3, MLKL, and PGAM5, remains to be determined. In addition, nanosecond pulsed electric fields and gemcitabine induce similar cell death pathways, particularly in breast cancer MCF-7 cells; however, the two agents exhibit different mechanisms of necrosis, most likely necroptosis, in breast cancer MDA-MB-231 cells [182]. Endoplasmic reticulum stress can also trigger RIPK1 kinase dependent necroptosis [183], and viruses can induce necroptosis in ovarian carcinoma cells and neuroblastoma cell lines [184-185].
Crosstalk between necroptosis and other forms of cancer cell death forms
Despite involving morphologically different and distinct molecular pathways, the pathways controlling different cell death modes involve complex interaction and crosstalk [186].
Induction of the mixed forms of death
Several studies have demonstrated that some drugs and kinase inhibitors, and heat stress, induce simultaneous apoptosis, necroptosis and autophagy in cancer cells [187-214] (Supplementary File 2). Furthermore, the combination of necroptotic inducers and standard treatments enhanced this form of cancer cell death. For example, gambogenic acid, one of the main components of Gamboge, and 5-Fu have synergistic effects on A549 cells, activating cell death through apoptotic and necroptotic mechanisms via the ROS-mitochondrial pathway [215]. Treatment of colorectal tumor cells with RT and hyperthermia also activates apoptosis and necroptosis [174]. These forms of cancer cell death highlight the interconnected nature of cell the involved pathways, and interdependence of the different modes of cancer cell death [22, 216].
Induction of apoptosis engages necroptosis
Some agents that are well-known inducers of apoptosis can also induce necroptosis under certain conditions. For example, staurosporine has been reported to trigger necroptosis in leukemia cells, and necroptosis was blocked by the RIPK1 inhibitor Nec-1 and MLKL inhibitor NSA [217]. The death receptor ligand TRAIL can also trigger necroptosis in colon and liver cancer cells at acidic extracellular PH [218-220]. Cancer cells shift from apoptosis to programmed necrosis or necroptosis when energy stores are depleted, such as during DNA repair by poly-ADP-ribose polymerase (PARP) activation, or switching of the extracellular PH in human HT29 and HepG2 cells [220-221]. However, Sosna et al. [222] have shown that necroptosis and PARP-1-mediated necrotic cell death follow two distinct routes to regulate necrosis. In addition, small molecule inhibitors that antagonize inhibitors of apoptotic proteins, such as second mitochondrial activator of caspases (smac) mimetics, were shown to cause necroptotic cell death in fibrosarcoma cells, breast cancer cells, and acute lymphoblastic leukemia, underscoring the potential clinical relevance of this therapeutic strategy [223-224], which is regulated by ROS [225]. Therefore, whether cancer cells undergo apoptosis or necroptosis following exposure to smac mimetics will be crucial for the implementation of therapy, and should be addressed in future studies.
Induction of necroptosis engages apoptosis
In contrast to the above-mentioned results, some agents inducing of necroptosis were also found to induce apoptosis under certain conditions. For example, shikomin and its analogues induced necroptosis, but in the presence of Nec-1, cells revert to apoptosis. The death mode switch is at least partially due to the conversion from mitochondrial inner membrane permeability to mitochondrial outer membrane permeability, which is associated with Bax translocation [226-227]. In addition, shikonin at low doses induces caspase dependent apoptosis, but at high doses induces necroptosis. Gene expression profiling revealed that 353 genes were significantly regulated by low dose shikonin and 85 genes by high dose shikonin. Among these genes, the transcription factor 3 and DNA damage inducible transcript 3 were highly expressed following low dose treatment, while high doses induced tumor necrosis factor expression. These findings provide novel information about the molecular mechanism of shikonin-induced apoptosis and necroptosis [228]. In combination with a previous report [16], these findings seem to support the notion that apoptosis and necroptosis may function as reciprocal backup mechanisms. Although the precise molecular mechanisms by which these agents induce cell death modes remain to be elucidated, these results indicate multiple pathways for the treatment of cancer.
Induction of autophagy engages necroptosis
Little is known about how autophagy is intertwined with necroptosis. However, some of the first evidence to show that autophagy could promote cell death came from a system that has gone on to become the best understood necroptosis pathway [229-232]. In this case, autophagy was shown to modulate necroptosis by selectively degrading the ROS scavenger enzyme catalase [233]. In another example, one piece of evidence suggests that autophagosomal membranes act as platforms for necrosome assembly, and serves as key sites to mediate necroptosis. For example, autophagosomes can act as key mediators of efficient necrosome formation, resulting in necroptotic cell death by Obatoclax (GX15-070), which is a compound that antagonizes Bcl-2 family proteins [152]. On the contrary, Bray et al. provided another example of the coordination between necroptosis and autophagy [234]. They found that concurrent mTOR inhibition by CCI-779 and inhibition of autophagosome maturation with chloroquine caused accumulation of autophagosomes that induced RIPK3-dependent and ROS-dependent necroptosis in renal cell carcinoma lines. RIPK1 may also degrade by autophagy. Overall, these data support the notion that autophagy can influence the fate of cells treated with compounds that induce necroptosis. However, the molecular underpinnings of this relationship remain largely elusive and somewhat controversial. Autophagy has been shown to induce [152, 229-233], suppress [117, 235-236] or not be involved in necroptosis [237]. Further work will be required to uncover the mechanistic ties, and to determine how these processes are controlled.
Necroptosis overcomes therapy resistance of cancer
Cancer resistance is a major obstacle limiting the efficacy of cancer therapy. Many lines of clinical and experimental evidence have demonstrated that an apoptotic defect is the most frequent cause of cancer resistance [23]. First, since the neoplastic process is driven by oncogenic mutations that increase tumor cell number via activating the cell cycle and/or inhibiting the normal apoptotic process, cancer cells are genetically predisposed to apoptotic resistance. Second, since conventional anticancer therapies, regardless of their targets and mechanisms, mostly induce apoptosis, therapy resistance via anti-apoptotic progression appears to be inevitable, and the clones with greater selective advantages against apoptosis are destined to dominate the total cancerous cell population, forming cancers in an ever more relentless cycle [238-239].
Apoptotic defects in cancers include over-expression of anti-apoptotic proteins, mutations of pro-apoptotic proteins and the loss of caspases [239]. The blockage of a single pathway does not entirely block cell death; however, it may provoke the cell to choose an alternative death pathway [240]. Therefore, targeting the “weak point” of cancers, especially apoptotic/drug resistant cancers, is critical for the success of cancer therapy. The necroptotic pathway might be an intrinsic “weak point” under these circumstances, and thus exploitation of this alternative cell death pathway may help bypass the blockade to destroy therapy-resistant cancers [241].
Generally speaking, apoptosis can overshadow necroptosis, implying that apoptotic death is the first choice in most settings and that necroptosis acts only as an alternative to ensure cell demise [242]. However, induction of necroptosis by a small molecular compound can circumvent cancer resistance. For example, Cisplatin-based chemotherapy is currently the standard treatment for locally advanced esophageal cancer, but cancer cells have evolved several strategies to evade apoptosis. Xu et al. [243] indicated that RIPK3 and the autocrine production of TNF-α contribute to cisplatin sensitivity by initiating necrosis when the apoptotic pathway is suppressed or absent in esophageal cancer cells. These findings provide new insight into the molecular mechanisms underlying cisplatin-induced necroptosis, and suggest that RIPK3 is a potential marker of cisplatin sensitivity in apoptosis-resistant and advanced esophageal cancer. The inhibition of glycogen synthase kinase 3 alpha (GSK3A) or glycogen synthase kinase 3 beta (GSK3B), whose silencing bypasses drug resistance due to the loss of p53, can abolish cell viability and cloning growth via necroptosis. Targeting GSK3B with/without CT may thus represent a novel strategy for the treatment of chemotherapy resistant tumors [244-246]. Meanwhile, a diphtheria based fusion toxin or smac mimetric, in addition to demethylating agents, synergistically trigger cell death in cancer cells and overcome apoptosis resistance by inducing necroptosis [247-249]. In addition, Trastuzumab, a humanized recombination monoclonal antibody that binds to the extracellular domain of Her2, can simultaneously induce necroptosis and overcome the resistance of tumor cells to antibody treatment [250]. These findings raise the possibility that induction of necroptosis may be considered an alternative choice for therapeutic resistant cancers.
Development of necroptotic inhibitors
Traditional chemotherapeutic agents are not efficient inducers of necroptosis, and more potent necroptotic pathway-specific drugs will be required to fully harness the power of necroptosis in anticancer therapy. We have reviewed the published literature on small molecule inhibition of necroptosis, and the development of specific necroptotic inducers, in combination with conventional therapeutic styles such as RT or CT, may be required to effectively treat cancers [251-252].
Nec-1
Small molecule compounds that inhibit programmed necrosis are not only effective tools for the study of necrotic cell death, but also have the potential to treatment for necroptosis. Nec-1 is the first necroptosis inhibitor to have been widely used in vitro or in animal models to study necroptosis. Nec-1 specifically inhibits necroptosis, but does not affect apoptosis and autophagy, and does not affect the general physiology of healthy cells, ATP levels, mitochondria membrane potential, plasma membrane integrity, cell shape and size, cell cycle distribution, proliferation, global mRNA expression, or intracellular ROS. The specificity of Nec-1-mediated inhibition of necroptosis has been well defined by extensive structure-activity relationship analyses, and Nec-1 was reported to allosterically inhibit the kinase activity of RIPK1, which is essential for death receptor triggered necroptosis, by interacting with its T-loop without affecting other functional domains [16]. Although many studies of necroptosis are exclusively based on experiments showing that Nec-1 inhibits cell death in the corresponding cell death model, these studies should be interpreted with caution as RIPK1 can regulate not only necroptotic but also apoptotic cell death, and therefore, Nec-1 might block apoptosis under specific conditions as well as ferroptosis except for necroptosis, in contrast to Nec-1s and ponatinib [253]. Furthermore, Linkermann et al. demonstrated that the in vivo effects of Nec-1 did not necessarily involve necrosis [254].
GSKs and NSA
Given that RIPK3 is involved in necroptosis, but not apoptosis, a RIPK3 inhibitor may be a more selective inhibitor of necroptosis. Necroptosis can be pharmacologically inhibited by RIPK3 inhibitors GSK-843, GSK-872, and GSK-840 [255-256]. RIPK3 silencing in cancer cells was reported to suppress the complex regulation of the apoptosis/necroptosis switch and NF-κB activation [257]. In addition, MLKL might also mediate signal transduction beyond RIPK3, as it is more widely expressed than RIPK3. It would therefore be interesting to identify the kinases with which MLKL interacts in the context of other signaling events [34]. MLKL expression, an independent prognostic biomarker in patients with early-stage resected pancreatic carcinoma, when low, is associated with a decrease in overall survival and recurrence-free survival in resected pancreatic carcinoma patients who receive adjuvant chemotherapy, even after accounting for adverse tumor characteristics and other known prognostic biomarkers [64]. Several studies reveal that NSA blocked necroptosis downstream of RIPK3 activation [29], and also targeted the N-terminal fragment of MLKL, and covalently modified MLKL through a Michael addition at a reactive amino acid residue cysteine. Therefore, NSA may represent a specific inhibitor of necroptosis. However, it should be noted that NSA does not work with murine MLKL, and currently lacks specificity.
Pazopanib and ponatinib
Drugs capable of inhibiting necroptosis may be useful in treating pathologies caused or aggravated by necroptotic cell death. For example, Fauster et al. performed a phenotypic screen for small-molecule inhibitors of TNF-alpha induced necroptosis in FADD deficient Jurkat cells using a representative panel of FDA approved drugs. Two structurally distinct kinase inhibitors, pazopanib and ponatinib, were found to be potent blockers of necroptosis, which do not protect from apoptosis [258]. Pazopanib and ponatinib abrogated phosphorylation of MLKL upon TNF-alpha induced necroptosis, indicating that both agents target a component upstream of MLKL. Importantly, ponatinib inhibited both RIPK1 and RIPK3, while pazopanib preferentially targeted RIPK1. Further studies will clarify the potential necroptosis-related clinical applications of these drugs which, given their potency in cellular assays and favorable pharmacological properties, could otherwise provide the basis for development of necroptosis inhibitors.
Others
In addition to Nec-1, GSKs, and NSA, a few other necroptotic inhibitors have been developed. Geserick et al. [259] reported that Dabrafenib, a V600E- or V600K mutated porto-oncogene serine/threonine protein kinase B-RAF, inhibited necroptosis in melanoma cells whenever RIPK3 is present. Wang et al. [260] reported that the histone deacetylase inhibitor vorinostat prevents TNF-α-induced necroptosis by regulating multiple signaling pathways, and suggested that histone deacetylase inhibition exerts its anti-inflammatory and cell protective effects by preventing necroptosis, an important process in inflammation and the elimination of cells. Onizawa et al. reported that necroptosis is inhibited by deubiquitinating enzyme A20, also known as tumor necrosis factor induced protein 3 (TNFAP3), and demonstrated that it prevents RIPK3-mediated necroptosis by blocking the ubiquitination of RIPK3 and formation of the RIPK1-RIPK3 complex [73, 261]. In addition, Carmina-Gutierrez et al. [262] reported that the propeptide of yeast cathepsin D inhibits necroptosis. Other necroptosis inhibitors such as Nec-7, which inhibits necroptosis independently of RIPK1, are also available [263-264].
The potential side effects triggered by necroptotic inhibitors in acute settings might differ from those of long-term anticancer treatment, and require careful evaluation. The identification of FDA approved drugs as new inhibitors of necroptosis, together with the elucidation of their mechanism of action, warrants a series of careful studies in animal models covering a wide variety of necroptosis associated pathologies. These studies may clarify the clinical potential of these necroptosis-related drugs which, given their potency in cellular assays and favorable pharmacological properties, could allow further development of clinically useful necroptosis inhibitors.
The resistant mechanism of necroptosis
Acquired or intrinsic resistance to necroptosis may be considered a major hindrance to therapeutic success in cancers. RIPK3 expression is likely repressed during cancer development or progression, by methylation of the genomic region near its transcriptional start site. Thus RIPK3 dependent activation of MLKL and downstream-programmed necroptosis during therapeutic death is largely repressed [265]. These results suggest that RIPK3 deficient cancer patients might benefit from agents inducing RIPK3 expression prior to treatment with conventional therapeutics. Seneviratne et al. [266] showed that defective DNA MMR system-induced genomic instability causes the truncation of an HGF gene promoter element, which reactivates the otherwise silenced HGF gene in colonic epithelial cells. This lead to autocrine HGF production and dysregulated Met signaling in colon cancer cells, thereby promoting their resistance to necroptosis. In addition, hypoxia inducible factor-1α and glucose transporters were recently reported to colocalize at peri-necrotic regions in human colorectal tumors [267-268]. Moriwaki et al. [252] further reported that expression of RIPK1 and RIPK3 was suppressed by hypoxia in human colon cancer tissues. These results suggest that glucose metabolism might confer anti-necrotic resistance to hypoxia stress. Malignant cells develop adaptive mechanisms to evade necrotic death caused by the depletion of oxygen and nutrients. Huang et al. [269] demonstrated a novel mechanism through which glycolytic pyruvate conferred resistance to RIP-dependent necroptosis in hypoxic colorectal carcinoma via mitochondrial superoxide scavenging. Understanding of how cells develop resistance to hypoxic necrosis through glucose and pyruvate may aid development of novel therapeutic targets for treating colorectal carcinoma.
Future directions
Recently, extensive studies have elucidated the molecular mechanisms by which necroptosis is regulated and the intricate crosstalk between different cell death modes. However, our understanding of the necroptosis pathway is still at an infant stage, and the execution of necroptosis remains unclear. We still lack a biomarker for the in vivo detection of necroptosis. Induction of necrosis may not only eliminate tumor cells directly, but may also invoke host innate immune responses to aid clearance of tumor cells. More experimental and clinical trials will be required to clarify the potential for necroptosis-targeted cancer therapy. Although the effects of therapy-induced necroptosis in cancer remain controversial, the hypothesis provides a novel perspective and may yield a new way to overcome therapy resistance in the treatment of cancer.
CONCLUSION
Since evasion of apoptosis represents a hallmark of human cancers, it follows that engagement of necroptosis may offer new opportunities for the development of novel treatments for apoptosis -resistant cancers. Since treatment resistance is currently the most challenging unsolved problem in oncology, therapeutic induction of necroptosis may pave an avenue for novel and more efficient treatment approaches. While a number of different anticancer compounds have been reported to trigger necroptosis in malignant cells, little is known about the precise molecular targets of these compounds. In conclusion, a better understanding of the molecular events that regulate necroptotic cell death in different types of human cancers is expected to provide exciting novel opportunities in the coming years for the therapeutic exploitation of cell death programs for the treatment of cancer. Further studies to identify novel biomarkers of necroptosis, and to develop tools for the precise characterization of necroptosis, distinct from other forms of programmed necrosis, in animal models and human disease samples, will provide crucial insight into the diagnosis, treatment, and monitoring of necroptosis-associated disease.
ACKNOWLEDGMENTS
We apologize to researchers whose studies we were unable to cite due to the length of this review. We are indebted to all our colleagues at the Laboratory of Cancer Cell Biology of Tianjin Medical University Cancer Institute & Hospital who provided assistance for this study. We also thank an anonymous referee for his/her very helpful comments, which remarkably improved the quality of this paper.
CONFLICTS OF INTEREST
We have no conflicts of interests. There is no copyrighted information in this article: No text, text boxes, figures, or tables in this article have been previously published or owned by another party.
FUNDING
This work was supported in part by the National Natural Science Foundation of China (No. 81301624, No. 81201754, and No. 81472797), the New Teacher Fund for Doctor Station, and the Ministry of Education (No. 20121202120014). No benefits in any form have been or will be received from a commercial party directly or indirectly related to the subject of this article.
Author contributions
MBM conducted the literature search, wrote and edited the manuscript, created the figures, and provided direction for the review as the senior author. HHW, YLC, and YYS assisted in the literature search as well as in the writing and editing of the manuscript. NGZ and LD assisted in the editing of the manuscript. ZQW assisted in the creation of the figures. ZYY, YL, and PW provided guidance in the revision stage. All authors have read and approved the manuscript.
REFERENCES
1. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med 2009; 361: 1570-1583.
2. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 2015; 22: 58-73.
3. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700-714.
4. Galluzzi L, Kepp O, Kroemer G. RIP kinases initiate programmed necrosis. J Mol Cell Biol 2009; 1: 8-10.
5. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK, Wu H. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012; 150: 339-350.
6. Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell 2008; 135: 1161-1163.
7. Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 2010; 17: 922-930.
8. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, et al. Nomenclature Committee on Cell Death 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16: 3-11.
9. Linkermann A, Green DR. Necroptosis. N Engl J Med 2014; 370: 455-465.
10. Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is proceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ 2016; 23: 253-260.
11. Yu X, Deng Q, Bode AM, Dong Z, Cao Y. The role of necroptosis, and alternative form of cell death, in cancer therapy. Expert Rev Anticancer Ther 2013; 13: 883-893.
12. Fulda S. Therapeutic exploitation of necroptosis for cancer therapy. Semin Cell Dev Biol 2014; 35: 51-56.
13. Cho YS, Park SY. Harness of programmed necroptosis for fighting against cancers. Biomes Ther 2014; 22: 167-175.
14. Fulda S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol Ther 2013; 14: 999-1004.
15. Jain MV, Paczulla AM, Klonisch T, Dimgba FN, Rao SB, Roberg K, Schweizer F, Lengerke C, Davoodpour P, Palicharla VR, Maddika S, Łos M. Interconnections between apoptosis, autophagic and necrotic pathways: implications for cancer therapy development. J Cell Mol Med 2013; 17: 12-29.
16. Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ 2016; 23: 748-756.
17. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646-674.
18. Hanahan D, Weinberg RA. Hallmarks of cancer. Cell 2000; 100: 57-70.
19. Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ 2012; 19: 75-86.
20. Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 1998; 187: 1477-1485.
21. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1: 489-495.
22. Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4: 313-321.
23. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-derived assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112-1123.
24. He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100-1111.
25. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, and energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332-336.
26. Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B, Gollnick H, Silke J, Leverkus M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol 2009; 187: 1037-1054.
27. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Häcker G, Leverkus M. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43: 449-463.
28. Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal 2010; 3: re4.
29. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213-227.
30. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA 2012; 109: 5322-5327.
31. Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, Oberst A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ 2014; 21: 1511-1521.
32. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013; 39: 443-453.
33. Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 2016; 23: 76-88.
34. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014; 16: 55-65.
35. Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 2014; 24: 105-121.
36. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54: 133-146.
37. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 2014; 7: 971-981.
38. Su L, Quade B, Wang H, Sun L, Wang X, Rizo J. A plug release mechanism for membrane permeation by MLKL. Structure 2014; 22: 1489-1500.
39. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012; 148: 228-243.
40. Takeda K, Komuro Y, Hayakawa T, Oguchi H, Ishida Y, Murakami S, Noguchi T, Kinoshita H, Sekine Y, Iemura S, Natsume T, Ichijo H. Mitochondrial phosphoglycerate mutase 5 uses alternate catalytic activity as a protein serine/threonine phosphatase to activate ASK1. Proc Natl Acad Sci USA 2009; 106: 12301-12305.
41. Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, Kidd G, Wakefield R, Frase S, Krautwald S, Linkermann A, Green DR. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep 2013; 5: 878-885.
42. Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O’Donnell CL, Gough PJ, Bertin J, Welsh RM, Chan FK. The mitochondrial phosphatase PGAM5 is dispensable for necroptosis but promotes inflammasome activation in macrophages. J Immunol 2016; 196: 407-415.
43. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181-190.
44. Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha -induced cell death by inducing c-FLIP (L) turnover. Cell 2006; 124: 601-613.
45. Silke J, Brink R. Regulation of TNFRSF and innate immune signaling complexes by TRAFs and cIAPs. Cell Death Differ 2010; 17: 35-45.
46. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAPs and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30: 689-700.
47. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 2006; 22: 245-257.
48. Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domain of A20 downregulate NF-kappaB signaling. Nature 2004; 430: 694-699.
49. Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sic STKE 2006; 2006: re13.
50. Fan Y, Yu Y, Shi Y, Sun W, Xie M, Ge N, Mao R, Chang A, Xu G, Schneider MD, Zhang H, Fu S, Qin J, Yang J. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha-and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J Biol Chem 2010; 285: 5347-5360.
51. Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, Giansanti P, Heck AJ, Dejardin E, Vandenabeele P, Bertrand MJ. NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell 2015; 60: 63-76.
52. Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996; 274: 787-789.
53. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature 2015; 517: 311-320.
54. Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693-703.
55. Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998; 8: 297-303.
56. Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 2013; 20: 1381-1392.
57. Legarda-Addison D, Hase H, O’Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB -independent cell-death checkpoint during TNF signaling. Cell Death Differ 2009; 16: 1279-1288.
58. Yang S, Wang B, Tang LS, Siednienko J, Callanan JJ, Moynagh PN. Pellino3 targets RIP1 and regulates the pro-apoptosis effects of TNF-α. Nat Commun 2013; 4: 2583.
59. Han J, Zhong CQ, Zhang DW. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol 2011; 12: 1143-1149.
60. Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernández-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevent RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011; 477: 330-334.
61. Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, et al. RIPK1 regulates RIPK3-MLKL-derived systemic inflammation and emergency hematopoiesis. Cell 2014; 157: 1175-1188.
62. Lin J, Li H, Yang M, Ren J, Huang Z, Han F, Huang J, Ma J, Zhang D, Zhang Z, Wu J, Huang D, Qiao M, Jin G, Wu Q, Huang Y, Du J, Han J. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep 2013; 3: 200-210.
63. Sato K, Li S, Gordon WC, He J, Liou GI, Hill JM, Travis GH, Bazan NG, Jin M. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J Neurosci 2013; 33: 17458-17468.
64. Colbert LE, Fisher SB, Hardy CW, Hall WA, Saka B, Shelton JW, Petrova AV, Warren MD, Pantazides BG, Gandhi K, Kowalski J, Kooby DA, El-Rayes BF, et al. Pronecrosis mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer 2013; 119: 3148-3155.
65. Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 promote TNF-induced apoptosis. Genes Dev 1999; 13: 2514-2526.
66. Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 2007; 19: 2056-2067.
67. O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol 2011; 13: 1437-1442.
68. Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 2011; 35: 572-582.
69. Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011; 471: 368-372.
70. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP (L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471: 363-367.
71. Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011; 471: 373-376.
72. Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. Identification of molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008; 135: 1311-1323.
73. Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, Whang MI, Advincula R, Agelidis A, Barrera J, Wu H, Burlingame A, Malynn BA, Zamvil SS, Ma A. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol 2015; 16: 618-627.
74. Peltzer N, Darding M, Walczak H. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol 2016; 26: 445-461.
75. Dondelinger Y, Darding M, Bertrand MJ, Walczak H. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol Life Sci 2016.
76. Yang Y, Hu W, Feng S, Ma J, Wu M. RIP3bata and RIP3r, two novel splice variants of receptor-interacting protein (RIP3), down-regulate RIP3-induced apoptosis. Biochem Biophys Res Commun 2005; 332: 181-187.
77. Kasof GM, Prosser JC, Liu D, Lorenzi MV, Gomes BC. The RIP-like kinase, RIP3, induce apoptosis and NF-kb nuclear translocation and localizes to mitochondria. FEBS Lett 2000; 473; 285-291.
78. Wu W, Liu P, Li J. Necroptosis: an emerging form of programmed cell death. Crit Rev Oncol Hematol 2012; 82: 249-258.
79. Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kB signaling. Cell 2006; 125: 665-677.
80. Alameda JP, Moreno-Maldonado R, Navarro M, Bravo A, Ramírez A, Page A, Jorcano JL, Fernández-Aceñero MJ, Casanova ML. An inactivating CYLD mutation promotes skin tumor progression by conferring enhanced proliferative, survival and angiogenic properties to epidermal cancer cells. Oncogene 2010; 29: 6522-6532.
81. Liu ZY, Wu B, Guo YS, Zhou YH, Fu ZG, Xu BQ, Li JH, Jing L, Jiang JL, Tang J, Chen ZN. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am J Cancer Res 2015; 5: 3174-3185.
82. Reed JC. Drug insight: cancer strategies based on restoration of endogenous cell death mechanisms. Nat Clin Prac Oncol 2006; 3: 388-398.
83. Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptosis cells: getting rid of the corpses. Mol Cell 2004; 14: 277-287.
84. Wagner BJ, Lindau D, Ripper D, Stierhof YD, Glatzle J, Witte M, Beck H, Keppeler H, Lauber K, Rammensee HG, Königsrainer A. Phagocytosis of dying tumor cells by human peritoneal mesothelial cells. J Cell Sci 2011; 124: 1644-1654.
85. Ravichandran KS. Beginning of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity 2011; 35: 445-455.
86. Ravichandran KS. Fand-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med 2010; 207: 1807-1817.
87. Munoz LE, Peter C, Herrmann M, Wesselborg S, Lauber K. Scent of dying cells: the role of attraction signals in the clearance of apoptotic cells and its immunological consequences. Autoimmune Rev 2010; 9: 425-430.
88. Peter C, Wesselborg S, Herrmann M, Lauber K. Dangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis 2010; 15: 1007-1028.
89. Shi Y, Mucsi AD, Ng G. Monosodium urate crystals in inflammation and immunity. Immunol Rev 2010; 233: 203-217.
90. Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, Herrmann M, Voll RE. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 2008; 205: 3007-3018.
91. Blume KE, Soeroes S, Keppeler H, Stevanovic S, Kretschmer D, Rautenberg M, Wesselborg S, Lauber K. Cleavage of annexin A1 by ADAM10 during secondary necrosis generates a monocytic “find-me” signal. J Immunol 2012; 188: 135-145.
92. Blume KE, Soeroes S, Waibel M, Keppeler H, Wesselborg S, Herrmann M, Schulze-Osthoff K, Lauber K. Cell surface externalization of annexin A1 as a failsafe mechanism preventing inflammatory responses during secondary necrosis. J Immunol 2009; 183: 8138-8147.
93. Caipl US, Kuenkele S, Voll RE, Beyer TD, Kolowos W, Heyder P, Kalden JR, Herrmann M. Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ 2001; 8: 327-334.
94. Bottcher A, Gaipl US, Furnrohr BG, Herrmann M, Girkontaite I, Kalden JR, Voll RE. Involvement of phosphatidylserine, alphavbeta3, CD14, CD36, and complement C1q in the phagocytosis of primary necrotic lymphocytes by macrophage. Arthritis Rheum 2006; 54: 927-938.
95. Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN, Haegeman G, D’Herde K, Vandenabeele P. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell 2004; 15: 1089-1100.
96. Ahrens S, Zelenay S, Sancho D, Hanč P, Kjær S, Feest C, Fletcher G, Durkin C, Postigo A, Skehel M, Batista F, Thompson B, Way M, Reis e Sousa C, Schulz O. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 2012; 36: 635-645.
97. Zhang JG, Czabotar PE, Policheni AN, Caminschi I, Wan SS, Kitsoulis S, Tullett KM, Robin AY, Brammananth R, van Delft MF, Lu J, O’Reilly LA, Josefsson EC, et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity 2012; 36: 646-657.
98. Sancho D, Joffre OP, Keller AM, Rogers NC, Martínez D, Hernanz-Falcón P, Rosewell I, Reis e Sousa C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009; 458: 899-903.
99. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010; 11: 889-896.
100. Steinman RM. Dendritic cells: understanding immunogenicity. Eur J Immunol 2007; 37 Suppl 1: S53-S60.
101. Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, Linkermann A, Virgin HW, Green DR. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 2016; 533: 115-119.
102. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 2013; 38: 209-223.
103. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Métivier D, Pichard E, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 2005; 202: 1691-1701.
104. Hirt UA, Leist M. Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ 2003; 10: 1156-1164.
105. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, Mendiboure J, Pignon JP, Jooste V, van Endert P, Ducreux M, Zitvogel L, Piard F, Kroemer G. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010; 29: 482-491.
106. Yatim N, Jusforgues-Saklani H, Orozco O, Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, Albert ML. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 2015; 350: 328-334.
107. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006; 6: 836-848.
108. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Métivier D, Larochette N, van Endert P, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13: 54-61.
109. Schmidt SV, Seibert S, Walch-Ruckheim B, Vicinus B, Kamionka EM, Pahne-Zeppenfeld J, Solomayer EF, Kim YJ, Bohle RM, Smola S. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget 2015; 6: 8635-8647. doi: 10.18632/oncotarget.3249.
110. Takemura R, Takaki H, Okada S, Shime H, Akazawa T, Oshiumi H, Matsumoto M, Teshima T, Seya T. PolyI:C-induced, TLR3/RIP3-dependent necroptosis backs up immune effector-mediated tumor elimination in vivo. Cancer Immunol Res 2015; 3: 902-914.
111. Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, Lerner RA, Otsuka M. Regulation of NKT cell-mediated immune responses to tumors and liver inflammation by mitochondrial PGAM5-Drp1 signaling. Nat Commun 2015; 6: 8371.
112. Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P, Garg AD, Leybaert L, Grooten J, et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep 2016; 15: 274-287.
113. Smola S. RIPK3-a predictive marker for personalized immunotherapy. Oncoimmunology 2015; 5: e1075695.
114. Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 2011; 477: 335-339.
115. Grivennikov SI, Greten FR, Karin M. Immunity inflammation, and cancer. Cell 2010; 140: 883-899.
116. Lu JV, Chen HC, Walsh CM. Necroptotic signaling in adaptive and innate immunity. Semin Cell Dev Biol 2014; 35: 33-39.
117. Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, Morrissette NS, Walsh CM. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA 2008; 105: 16677-16682.
118. Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med 2011; 208: 633-641.
119. Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, Wakeham A, Bouchard D, Yeh WC, McGlade JC, Ohashi PS, Hakem R. Essential role for caspase-8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev 2003; 17: 883-895.
120. Osborn SL, Diehl G, Han SJ, Xue L, Kurd N, Hsieh K, Cado D, Robey EA, Winoto A. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci USA 2010; 107: 13034-13039.
121. Newton K, Harris AW, Bath ML, Smith KG, Strasser A. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J 1998; 17: 706-718.
122. Walsh CW, Web BG, Chinnaiyan AM, O’Rourke K, Dixit VM, Hedrick SM. A role for FADD in T cell activation and development. Immunity 1998; 8: 439-449.
123. Degterev A, Maki JL, Yuan J. Activity and specificity of necrostatin-1, small molecule inhibitor of RIP1 kinase. Cell Death Differ 2013; 20: 366.
124. Ch’en IL, Beisner DR, Degterev A, Lynch C, Yuan J, Hoffmann A, Hedrick SM. -mediated T cell expansion regulated by parallel pathways of death. Proc Natl Acad Sci USA 2008; 105: 17463-17468.
125. Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-CASPASE-8-cFLIP (L) complex. Cell Rep 2012; 1: 401-407.
126. Lu JV, Weist BM, van Raam BJ, Marro BS, Nguyen LV, Srinivas P, Bell BD, Luhrs KA, Lane TE, Salvesen GS, Walsh CM. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc Natl Acad Sci USA 2011; 108: 15312-15317.
127. Gamrekelashvili J, Kruger C, von Wasielewski R, Hoffmann M, Huster KM, Busch DH, Manns MP, Korangy F, Greten TF. Necrotic tumor cell death in vivo impairs tumor-specific immune responses. J Immunol 2007; 178: 1573-1580.
128. Cho Y, McQuade T, Zhang H, Zhang J, CHan FK. RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS One 2011; 6: e23209.
129. Szymczak-Workman AL, Delgoffe GM, Green DR, Vignali DA. Cutting edge: regulatory T cells do not mediate suppression via programmed cell death pathways. J Immunol 2011; 187: 4416-4420.
130. Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, Chen H, Dong H, Zhu Y, Miao K, Xu W, Li J. Dysregulation of TNFα-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia 2012; 26: 1293-1300.
131. Chaabane W, User SD, EI-Gazzah M, Jaksik R, Sajjadi E, Rzeszowska-Wolny J, Los MJ. Autophagy, apoptosis, mitoptosis and necrosis: interdependence between those pathways and effects on cancer. Arch Immunol Ther Exp 2013; 61: 43-58.
132. Tabata K, Hamano A, Akihisa T, Suzuki T. Kuguaglycoside C, a constituent of momordica charantia, induces caspase-independent cell death of neuroblastoma cells. Cancer Sci 2012; 103: 2153-2158.
133. Li N, He Y, Wang L, Mo C, Zhang J, Zhang W, Li J, Liao Z, Tang X, Xiao H. D-galactose induces necroptosis cell death in neuroblastoma cell line. J Cell Biochem 2011; 112: 3834-3844.
134. Wang G, Wang JJ, Yang GY, Du SM, Zeng N, Li DS, Li RM, Chen JY, Feng JB, Yuan SH, Ye F. Effects of quercetin nanoliposomes on C6 glioma cells through induction of type III programmed cell death. Int J Nanomedicine 2012; 7: 271-280.
135. Hernandez-Breijo B, Monserrat J, Ramirez-Rubio S, Cuevas EP, Vara D, Díaz-Laviada I, Fernández-Moreno MD, Román ID, Gisbert JP, Guijarro LG. Preclinical evaluation of azathioprine plus buthionine sulfoximine in the treatment of human hepatocarcinoma and colon carcinoma. World J Gastroenterol 2011; 17: 3899-3911.
136. Baba N, Higashi Y, Kanekura T. Japanese black vinegar “Izumi” inhibits the proliferation of human squamous cell carcinoma cells via necroptosis. Nutr Cancer 2013; 65: 1093-1097.
137. Zhang LY, Wu YL, Gao XH, Guo F. Mitochondrial protein cyclophilin-D-mediated programmed necrosis attributes to berberine-induced cytotoxicity in cultured prostate cancer cells. Biochem Biophys Res Commun 2014; 450: 697-703.
138. Voigt S, Philipp S, Davarnia P, Winoto-Morbach S, Röder C, Arenz C, Trauzold A, Kabelitz D, Schütze S, Kalthoff H, Adam D. TRAIL-induced programmed necrosis as a novel approach to eliminate tumor cells. BMC cancer 2014; 14: 74.
139. Park EJ, Min KJ, Lee TJ, Yoo YH, Kim YS, Kwon TK. β-lapachone induces programmed necrosis through the RIP1-PARP-AIF-dependent pathway in human hepatocellular carcinoma SK-Hep1 cells. Cell Death Dis 2014; 5: e1230.
140. Huang X, Dong Y, Bey EA, Kilgore JA, Bair JS, Li LS, Patel M, Parkinson EI, Wang Y, Williams NS, Gao J, Hergenrother PJ, Boothman DA. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res 2012; 72: 3038-3047.
141. Sansone C, Braca A, Ercolesi E, Romano G, Palumbo A, Casotti R, Francone M, Ianora A. Diatom-derived polyunsaturated aldehydes activate cell death in human cancer cell lines but not normal cells. PLoS One 2014; 9: e101220.
142. Wang HY, Zhang B. Cobalt chloride induces necroptosis in human colon cancer HT-29 cells. Asian Pac J Cancer Prev 2015; 16: 2569-2574.
143. Philipp S, Sosna J, Plenge J, Kalthoff H, Adam D. Homoharringtonine, a clinically approved anti-leukemia drug, sensitizes tumor cells for TRAIL-induced necroptosis. Cell Commun Signal 2015; 13: 25.
144. Yu X, Deng Q, Li W, Xiao L, Luo X, Liu X, Yang L, Peng S, Ding Z, Feng T, Zhou J, Fan J, Bode AM, Dong Z, Liu J, Cao Y. Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFα and ROS production. Oncotarget 2015; 6: 1995-2008. doi: 10.18632/oncotarget.3038.
145. Zou X, Zhang M, Sun Y, Zhao S, Wei Y, Zhang X, Jiang C, Liu H. Inhibitory effects of 3-bromopyruvate in human nasopharyngeal carcinoma cells. Oncol Rep 2015; 34: 1895-1904.
146. Luo Y, Roy M, Xiao X, Sun S, Liang L, Chen H, Fu Y, Sun Y, Zhu M, Ye M, Liu J. Lycorine induces programmed necrosis in the multiple myeloma cell line ARH-77. Tumor Biol 2015; 36: 2937-2945.
147. Xie X, Zhao Y, Ma CY, Xu XM, Zhang YQ, Wang CG, Jin J, Shen X, Gao JL, Li N, Sun ZJ, Dong DL. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br J Pharmacol 2015; 172: 3929-3943.
148. Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, Zeng H, Zuo D, Hua Y, Cai Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013; 13: 580.
149. Huang C, Luo Y, Zhao J, Yang F, Zhao H, Fan W, Ge P. Shikonin kills glioma cells through necroptosis mediated by RIP1. PLoS One 2013; 8: e66326.
150. Shahsavari Z, Karami-Tehrani F, Salami S, Ghasemzadeh M. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: necroptosis as a desperate programmed suicide pathway. Tumor Biol 2015.
151. Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther 2007; 6: 1641-1649.
152. Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ 2013; 20: 1161-1173.
153. Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, Schäfer BW, Schrappe M, Stanulla M, Bourquin JP. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest 2010; 120: 1310-1323.
154. Rizzi F, Naponelli V, Silva A, Modernelli A, Ramazzina I, Bonacini M, Tardito S, Gatti R, Uggeri J, Bettuzzi S. Polyphenon E(R), a standardized green tea extract, induces endoplasmic reticulum stress, leading to death of immortalized PNT1a cells by anoikis and tumorigenic PC3 by necroptosis. Carcinogenesis 2014; 35: 828-839.
155. Wu M, Jiang Z, Duan H, Sun L, Zhang S, Chen M, Wang Y, Gao Q, Song Y, Zhu X, Zhang L. Deoxypodophyllotoxin triggers necroptosis in human non-small cell lung cancer NCI-H460 cells. Biomed Pharmacother 2013; 67: 701-706.
156. Jiang Y, Shan S, Chi L, Zhang G, Gao X, Li H, Zhu X, Yang J. Methyl methanesulfonate induces necroptosis in human lung adenoma A549 cells through the PIG-3-reactive oxygen species pathway. Tumor Biol 2016; 37: 3785-3795.
157. Lin W, Tongyi S. Role of Bax/Bcl-2 family members in green tea polyphenol induced necroptosis of p53-deficient Hep3B cells. Tumor Biol 2014; 35: 8065-8075.
158. Zhang H, Zhong C, Shi L, Guo Y, Fan Z. Granulysin induces cathepsin B release from lysosomes of target tumor cells to attack mitochondrial through processing of bid leading to necroptosis. J Immunol 2009; 182: 6993-7000.
159. Wallenberg M, Misra S, Wasik AM, Marzano C, Bjornstedt M, Gandin V, Fernandes AP. Selenium induces a multi-targeted cell death process in addition to ROS formation. J Cell Mol Med 2014; 18: 671-684.
160. Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ, Selvam SP, Senkal CE, Garrett-Mayer E, De Palma RM, et al. Sphingosine analogue drug FTY720 targets 12PP2A/SET and mediates lung tumor suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med 2013; 5: 105-121.
161. Zec M, Srdic-Rajic T, Krivokuca A, Jankovic R, Todorovic T, Andelkovic K, Radulovic S. Novel selenosemicarbazone metal complexes exert anti-tumor effect via alternative, caspase-independent necroptotic cell death. Med Chem 2014; 10: 759-771.
162. Tufo G, Jones AW, Wang Z, Hamelin J, Tajeddine N, Esposti DD, Martel C, Boursier C, Gallerne C, Migdal C, Lemaire C, Szabadkai G, Lemoine A, Kroemer G, Brenner C. The protein disulfide isomerases PDIA4 and PDIA6 mediate resistance to cisplatin-induced cell death in lung adenocarcinoma. Cell Death Differ 2014; 21: 685-695.
163. Coupienne I, Fettweis G, Rubio N, Agostinis P, Piette J. 5-ALA-PDT induces RIP3-dependent necrosis in glioblastoma. Photochem Photobiol Sci 2011; 10: 1868-1878.
164. He W, Wang Q, Srinivasan B, Xu J, Padilla MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, Xing C, Lin Y. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene 2014; 33: 3004-3013.
165. Qiu X, Klausen C, Cheng JC, Leung PC. CD40 ligand induces RIP1-dependent, necroptosis-like cell death in low-grade serous but not serous borderline ovarian tumor cells. Cell Death Dis 2015; 6: e1864.
166. Kaku Y, Touchiya A, Kanno T, Nakano T, Nishizaki T. Diarachidonoylphosphoethanolamine induces necrosis/necroptosis of malignant pleural mesothelioma cells. Cell Signal 2015; 27: 1713-1719.
167. Deeraksa A, Pan J, Sha Y, Liu XD, Eissa NT, Lin SH, Yu-Lee LY. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene 2013; 32: 2973-2983.
168. Liu X, Chhipa RR, Nakano I, Dasgupta B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Ther 2014; 13: 596-605.
169. Pasupuleti N, Leon L, Carraway KL 3rd, Gorin F. 5-Benzylglycinyl-amiloride kills proliferating and nonproliferating malignant glioma cells through caspase-independent necroptosis mediated by apoptosis-inducing factor. J Pharmacol Exp Ther 2013; 344: 600-615.
170. Leon LJ, Pasupuleti N, Gorin F, Carraway KL 3rd. A cell-permeant amiloride derivative induces caspase-independent, AIF-mediated programmed necrotic death of breast cancer cells. PLoS One 2013; 8: e63038.
171. Locatelli SL, Cleris L, Stirparo GG, Tartari S, Saba E, Pierdominici M, Malorni W, Carbone A, Anichini A, Carlo-Stella C. BIM upregulation and ROS-dependent necroptosis mediate the antitumor effects of the HDACi Givinostat and Sorafenib in Hodgkin lymphoma cell line xenografts. Leukemia 2014; 28: 1861-1871.
172. Kharaziha P, Chioureas D, Baltatzis G, Fonseca P, Rodriguez P, Gogvadze V, Lennartsson L, Björklund AC, Zhivotovsky B, Grandér D, Egevad L, Nilsson S, Panaretakis T. Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget 2015; 6: 37066-37082. doi: 10.18632/oncotarget.5797.
173. Nehs MA, Lin CI, Kozono DE, Whang EE, Cho NL, Zhu K, Moalem J, Moore FD Jr, Ruan DT. Necroptosis is a novel mechanism of radiation induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery 2011; 150: 1032-1039.
174. Mantel F, Frey B, Haslinger S, Schildkopf P, Sieber R, Ott OJ, Lödermann B, Rödel F, Sauer R, Fietkau R, Gaipl US. Combination of ionising irradiation and hyperthermia activates programmed apoptotic and necrotic cell death pathways in human colorectal carcinoma cells. Strahlenther Onkol 2010; 186: 587-599.
175. Schildkopf P, Frey B, Mantel F, Ott OJ, Weiss EM, Sieber R, Janko C, Sauer R, Fietkau R, Gaipl US. Application of hyperthermia in addition to ionizing irradiation fosters necrotic cell death and HMGB1 release of colorectal tumor cells. Biochem Biophys Res Commun 2010; 391: 1014-1020.
176. Das A, McDonald DG, Dixon-Mah YN, Jacqmin DJ, Samant VN, Vandergrift WA 3rd, Lindhorst SM, Cachia D, Varma AK, Vanek KN, Banik NL, Jenrette JM 3rd, Raizer JJ, Giglio P, Patel SJ. RIP1 and RIP3 complex regulates radiation-induced programmed necrosis in glioblastoma. Tumour Biol 2015; 37: 7525-7534.
177. Harberts E, Fishelevich R, Liu J, Atamas SP, Gaspari AA. MyD88 mediates the decision to die by apoptosis or necroptosis after UV irradiation. Innate Immun 2014; 20: 529-539.
178. Oliver Metzig M, Fuchs D, Tagscherer KE, Grone HJ, Schirmacher P, Roth W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-α-dependent necroptosis driven by RIP1 kinase and NF-κB. Oncogene 2015.
179. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010; 11: 597-610.
180. Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ. Biological functions of microRNAs: a review. J Physiol Biochem 2011; 67: 129-139.
181. Wang K, Liu F, Zhou LY, Ding SL, Long B, Liu CY, Sun T, Fan YY, Sun L, Li PF. miR-847 regulates myocardial necrosis by targeting caspase-8. Cell Death Dis 2013; 4: e709.
182. Wu S, Guo J, Wei W, Zhang J, Fang J, Beebe SJ. Enhanced breast cancer therapy with nsPEFs and low concentrations of gemcitabine. Cancer Cell Int 2014; 14: 98.
183. Saveljeva S, Mc Laughlin SL, Vandenabeele P, Samali A, Bertrand MJ. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis 2015; 6: e1587.
184. Whilding LM, Archibald KM, Kulbe H, Balkwill FR, Oberg D, McNeish IA. Vaccinia virus induces programmed necrosis in ovarian cancer cells. Mol Ther 2013; 21: 2074-2086.
185. Nomura M, Ueno A, Saga K, Fukuzawa M, Kaneda Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res 2014; 74: 1056-1066.
186. Lalaoui N, Lindqvist LM, Sandow JJ, Ekert PG. The molecular relationships between apoptosis, autophagy and necroptosis. Semin Cell Dev Biol 2015; 39: 63-69.
187. Urtishak KA, Edwards AY, Wang LS, Hudome A, Robinson BW, Barrett JS, Cao K, Cory L, Moore JS, Bantly AD, Yu QC, Chen IM, Atlas SR, et al. Potent obatoclax cytotoxicity and activation of triple death mode killing across infant acute lymphoblastic leukemia. Blood 2013; 121: 2689-2703.
188. Guido C, Panza S, Santoro M, Avena P, Panno ML, Perrotta I, Giordano F, Casaburi I, Catalano S, De Amicis F, Sotgia F, Lisanti MP, Andò S, Aquila S. Estrogen receptor beta (ERbeta) produces autophagy and necroptosis in human seminoma cell line through the binding of the Sp1 on the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) promoter gene. Cell Cycle 2012; 11: 2911-2921.
189. Horita H, Frankel AE, Thorburn A. Acute myeloid leukemia-targeted toxin activates both apoptotic and necroptotic death mechanisms. PLoS One 2008; 3: e3909.
190. Jang MS, Lee SJ, Kang NS, Kim E. Cooperative phosphorylation of FADD by Aur-A and Plk1 in response to taxol triggers both apoptotic and necrotic cell death. Cancer Res 2011; 71: 7207-7215.
191. Hammerova J, Uldrijan S, Taborska E, Vaculova AH, Slaninova I. Necroptosis modulated by autophagy is a predominant form of melanoma cell death induced by sanguilutine. Biol Chem 2012; 393: 647-658.
192. Zhang L, Wang H, Ding K, Xu J. FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicol Letter 2015; 236: 43-59.
193. Huang H, Xiao T, He L, Ji H, Liu XY. Interferon-β-armed oncolytic adenovirus induces both apoptosis and necroptosis in cancer cells. Acta Biochim Biophys Sin (Shanghai) 2012; 44: 737-745.
194. Yang C, Oh JS, Yoo SH, Lee JS, Yoon YG, Oh YJ, Jang MS, Lee SY, Yang J, Lee SH, Kim HY, Yoo YH. The targeted inhibition of mitochondrial Hsp90 overcomes the apoptosis resistance conferred by Bcl-2 in Hep3B cells via necroptosis. Toxicol Appl Pharmacol 2013; 266: 9-18.
195. Ramirez-Labrada A, Lopez-Royuela N, Jarauta V, Galán-Malo P, Azaceta G, Palomera L, Pardo J, Anel A, Marzo I, Naval J. Two death pathways induced by sorafenib in myeloma cells: puma-mediated apoptosis and necroptosis. Clin Transl Oncol 2015; 17: 121-132.
196. Deng Q, Yu X, Xiao L, Hu Z, Luo X, Tao Y, Yang L, Liu X, Chen H, Ding Z, Feng T, Tang Y, Weng X, Gao J, Yi W, Bode AM, Dong Z, Liu J, Cao Y. Neoalbaconol induces energy depletion and multiple cell death in cancer cells by targeting PDK1-PI3-K/Akt signaling pathway. Cell Death Dis 2013; 4: e804.
197. Thompson SM, Callstrom MR, Butters KA, Knudsen B, Grande JP, Roberts LR, Woodrum DA. Heat stress induced cell death mechanisms in hepatocytes and hepatocellular carcinoma: in vitro and in vivo study. Lasers Surg Med 2014; 46: 290-301.
198. Mouratidis PX, Rivens I, Ter Haar G. A study of thermal dose-induced antophagy, apoptosis and necroptosis in colon cancer cells. Int J Hyperthermia 2015; 31: 476-488.
199. Yan S, Yang X, Chen T, Xi Z, Jiang X. The PPARγ agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther 2014; 21: 188-193.
200. Yamanaka K, Urano Y, Takabe W, Saito Y, Noguchi N. Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acylCoA: cholesterol acyltransferase 1. Cell Death Dis 2014; 5: e990.
201. Pan Q, Huang Y, Chen L, Gu J, Zhou X. SMAC-armed vaccinia virus induces both apoptosis and necroptosis and synergizes the efficiency of vinblastine in HCC. Hum Cell 2014; 27: 162-171.
202. Xu B, Shu Y, Liu P. INF-γ sensitizes head and neck squamous cell carcinoma cells to chemotherapy-induced apoptosis and necroptosis through up-regulation of Egr-1. Histol Histopathol 2014; 29: 1437-1443.
203. Melo-Lima S, Celeste Lopes M, Mollinedo F. Necroptosis is associated with low procaspase-8 and active RIPK1 and -3 in human glioma cells. Oncoscience 2014; 1: 649-664. doi: 10.18632/oncoscience.89.
204. Shahsavari Z, Karami-Tehrani F, Salami S. Shikonin induced necroptosis via reactive oxygen species in the T-47D breast cancer cell line. Asian Pac J Cancer Prev 2015; 16: 7261-7266.
205. Wada N, Kawano Y, Fujiwara S, Okuno Y, Kikukawa Y, Endo S, Nishimura N, Ueno N, Mitsuya H, Hata H. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int J Oncol 2015; 46: 963-972.
206. Miki Y, Akimoto J, Moritake K, Hironaka C, Fujiwara Y. Photodynamic therapy using talaporfin sodium induces concentration-dependent programmed necroptosis in human glioblastoma T98G cells. Lasers Med Sci 2015; 30: 1739-1745.
207. Kaku Y, Tsuchiya A, Kanno T, Nakano T, Nishizaki T. Diarachidonoylphosphoethanolamine induces necrosis/necroptosis of malignant pleural mesothelioma cells. Cell Signal 2015; 27: 1713-1719.
208. Longato GB, Fiorito GF, Vendramini-Costa DB, de Oliveira Sousa IM, Tinti SV, Ruiz AL, de Almeida SM, Padilha RJ, Foglio MA, de Carvalho JE. Different cell death responses induced by eupomatenoid-5 in MCF-7 and 786-0 tumor cell lines. Toxicol In Vitro 2015; 29: 1026-1033.
209. De Ford C, Ulloa JL, Catalan CA, Grau A, Martino VS, Muschietti LV, Merfort I. The sesquiterpene lactone polymatin B from Smallanthus sonchifolius induces different cell death mechanisms in three cancer cell lines. Phytochemistry 2015; 117: 332-339.
210. Hirsch B, von der Wall E, Hummel M, Durkop H. RIP1 expression is necessary for CD30-mediated cell death induction in anaplastic large-cell lymphoma cells. Lab Invest 2013; 93: 677-689.
211. Laukens B, Jennewein C, Schenk B, Vanlangenakker N, Schier A, Cristofanon S, Zobel K, Deshayes K, Vucic D, Jeremias I, Bertrand MJ, Vandenabeele P, Fulda S. Smac mimetic bypasses apoptosis resistance in FADD- or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia 2011; 13: 971-979.
212. Radogna F, Cerella C, Gaigneaux A, Christov C, Dicato M, Diederich M. Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene 2015.
213. Jin J, Jin X, Qian C, Ruan Y, Jiang H. Signaling network of OSW-1-induced apoptosis and necroptosis in hepatocellular carcinoma. Mol Med Rep 2013; 7: 1646-1650.
214. Kaku Y, Tsuchiya A, Kanno T, Nishizaki T. HUHS1015 induces necroptosis and caspase-independent apoptosis of MKN28 human gastric cancer cells in association with AMID accumulation in the nucleus. Anticancer Agents Med Chem 2015; 15: 242-247.
215. Su J, Cheng H, Zhang D, Wang M, Xie C, Hu Y, Chang HC, Li Q. Synergistic effects of 5-fluorouracil and gambogenic acid on A549 cells: activation of cell death caused by apoptotic and necroptotic mechanisms via the ROS-mitochondria pathway. Biol Pharm Bull 2014; 37: 1259-1268.
216. Chaabane W, User SD, EI-Gazzah M, Jaksik R, Sajjadi E, Rzeszowska-Wolny J, Los MJ. Autophagy, apoptosis, mitoptosis and necrosis: interdependence between those pathways and effects on cancer. Arch Immunol Ther Exp (Warsz) 2013; 61: 43-58.
217. Dunai ZA, Imre G, Barna G, Korcsmaros T, Petak I, Bauer PI, Mihalik R. Staurosporine induces necroptotic cell death under caspase-compromised conditions in U937 cells. PLoS One 2012; 7: e41945.
218. Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, Takahashi N, Sergent O, Lagadic-Gossmann D, Vandenabeele P, Samson M, Dimanche-Boitrel MT. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 2012; 19: 2003-2014.
219. Meurette O, Huc L, Rebillard A, Le Moigne G, Lagadic-Gossmann D, Dimanche-Boitrel MT. TRAIL (TNF-related apoptosis-inducing ligand) induces necrosis-like cell death in tumor cells at acidic extracellular pH. Ann NY Acad Sci 2005; 1056: 379-387.
220. Meurette O, Rebillard A, Huc L, Le Moigne G, Merino D, Micheau O, Lagadic-Gossmann D, Dimanche-Boitrel MT. TRAIL induces receptor-interacting protein 1-dependent and caspase-dependent necrosis-like cell death under acidic extracellular conditions. Cancer Res 2007; 67: 218-226.
221. Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, Herceg Z, Wang ZQ, Schulze-Osthoff K. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell 2002; 13: 978-988.
222. Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P, Herdegen T, Linkermann A, Rittger A, Chan FK, Kabelitz D, Schütze S, Adam D. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci 2014; 71: 331-348.
223. Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, Vucic D, Fulda S, Vandenabeele P, Bertrand MJ. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ 2011; 18: 656-665.
224. Jin G, Lan Y, Han F, Sun Y, Liu Z, Zhang M, Liu X, Zhang X, Hu J, Liu H, Wang B. Smac mimetic-induced caspase-independent necroptosis requires RIP1 in breast cancer. Mol Med Rep 2016; 13: 359-366.
225. Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015; 34: 5796-5806.
226. Han W, Xie J, Li J, Liu Z, Hu X. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis 2009; 14: 674-686.
227. Han W, Xie J, Fang Y, Wang Z, Pan H. Nec-1 enhances shikonin-induced apoptosis in leukemia cells by inhibition of RIP-1 and ERK1/2. Int J Mol Sci 2012; 13: 7212-7225.
228. Piao JL, Cui ZG, Furusawa Y, Ahmed K, Rehman MU, Tabuchi Y, Kadowaki M, Kondo T. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: necroptosis as a desperate programmed suicide pathway. Chem Biol Interact 2013; 205: 119-127.
229. Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304: 1500-1502.
230. Fitzwalter B, Thorburn A. Recent insight into cell death and autophagy. FEBS J 2015; 282: 4279-4288.
231. Lu JV, Walsh CM. Metabolism and autophagy in the immune system: programmed necrosis and autophagy in immune function. Immunol Rev 2012; 249: 205-217.
232. Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 2013; 1833: 3448-3459.
233. Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 2006; 103: 4952-4957.
234. Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ 2013; 20: 1161-1173.
235. Bray K, Mathew R, Lau A, Kamphorst JJ, Fan J, Chen J, Chen HY, Ghavami A, Stein M, DiPaola RS, Zhang D, Rabinowitz JD, White E. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One 2012; 7: e41831.
236. Farkas T, Daugaard M, Jaattela M. Identification of Small Molecule Inhibitors of Phosphatidylinositol 3-Kinase and Autophagy. J Biol Chem 2011; 286: 38904-38912.
237. Osborn SL, Diehl G, Han SJ, Xue L, Kurd N, Hsieh K, Cado D, Robey EA, Winoto A. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci USA 2010; 107: 13034-13039.
238. Hu X, Han W, Li L. Targeting the weak point of cancer by induction of necroptosis. Autophagy 2007; 3: 490-492.
239. Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, Yang H, Samadi AK, Russo GL, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol 2015; 35 Suppl: S78-S103.
240. Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE 2006; pe44.
241. McCabe KE, Bacos K, Lu D, Delaney JR, Axelrod J, Potter MD, Vamos M, Wong V, Cosford ND, Xiang R, Stupack DG. Triggering necroptosis in cisplatin and IAP antagonist-resistant ovarian carcinoma. Cell Death Dis 2014; 5: e1496.
242. Krysko DV, Vanden Berghe T, D’Herde K, Vandenabeele P. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods 2008; 44: 205-221.
243. Xu Y, Lin Z, Zhao N, Zhou L, Liu F, Cichacz Z, Zhang L, Zhan Q, Zhao X. Receptor interactive protein kinase 3 promotes Cisplatin-triggered necrosis in apoptosis-resistant esophageal squamous cell carcinoma cells. PLoS One 2014; 9: e100127.
244. Grassilli E, Narloch R, Federzoni E, Ianzano L, Pisano F, Giovannoni R, Romano G, Masiero L, Leone BE, Bonin S, Donada M, Stanta G, Helin K, Lavitrano M. Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin Cancer Res 2013; 19: 3820-3831.
245. Grassilli E, Ianzano L, Bonomo S, Missaglia C, Cerrito MG, Giovannoni R, Masiero L, Lavitrano M. GSK3A is redundant with GSK3B in modulating drug resistance and chemotherapy-induced necroptosis. PLoS One 2014; 9: e100947.
246. Kwak CW, Son YM, Gu MJ, Kim G, Lee IK, Kye YC, Kim HW, Song KD, Chu H, Park BC, Lee HK, Yang DC, Sprent J, Yun CH. A bacterial metabolite, compound K, induces programmed necrosis in MCF-7 cells via GSK3β. J Microbiol Biotechnol 2015; 25: 1170-1176.
247. Steinhart L, Belz K, Fulda S. Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis 2013; 4: e802.
248. Chromik J, Safferthal C, Serve H, Fulda S. Smac mimetic primes apoptosis-resistant acute myeloid leukaemia cells for cytarabine-induced cell death by triggering necroptosis. Cancer Lett 2014; 344: 101-109.
249. Xuan Y, Hu X. Naturally-occurring shikonin analogues-a class of necroptotic inducers that circumvent cancer drug resistance. Cancer Lett 2009; 274: 233-242.
250. Hu J, Liu X, Hughes D, Esteva FJ, Liu B, Chandra J, Li S. Herceptin conjugates linked by EDC boost direct tumor cell death via programmed tumor cell necrosis. PLoS One 2011; 6: e23270.
251. Degterev A, Linkermann A. Generation of small molecules to interfere with regulated necrosis. Cell Mol Life Sci 2016.
252. Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis 2015; 6: e1636.
253. Vandenabeele P, Grootjans S, Callewaert N, Takahashi N. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ 2013; 20: 185-187.
254. Linkermann A, Heller JO, Prókai A, Weinberg JM, De Zen F, Himmerkus N, Szabó AJ, Bräsen JH, Kunzendorf U, Krautwald S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol 2013; 24: 1545-1557.
255. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 2013; 288: 31268-31279.
256. Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, Shen YY, Chen Y, Xiong B, Yang CH, Ding J, Miao ZH. The B-Raf (V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 2014; 5: e1278.
257. Nugues AL, EI Bouazzati H, Hetuin D, Berthon C, Loyens A, Bertrand E, Jouy N, Idziorek T, Quesnel B. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis 2014; 5: e1384.
258. Fauster A, Rebsamen M, Huber KV, Bigenzahn JW, Stukalov A, Lardeau CH, Scorzoni S, Bruckner M, Gridling M, Parapatics K, Colinge J, Bennett KL, Kubicek S, Krautwald S, Linkermann A, Superti-Furga G. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis 2015; 6: e1767.
259. Geserick P, Wang J, Schilling R, Horn S, Harris PA, Bertin J, Gough PJ, Feoktistova M, Leverkus M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis 2015; 6: e1884.
260. Wang D, Zhao M, Chen G, Cheng X, Han X, Lin S, Zhang X, Yu X. The histone deacetylase inhibitor vorinostat prevents TNFα-induced necroptosis by regulating multiple signaling pathways. Apoptosis 2013; 18: 1348-1362.
261. Gurung P, Man SM, Kanneganti TD. A20 is a regulator of necroptosis. Nat Immunol 2015; 16: 596-597.
262. Carmona-Gutierrez D, Bauer MA, Ring J, Knauer H, Eisenberg T, Büttner S, Ruckenstuhl C, Reisenbichler A, Magnes C, Rechberger GN, Birner-Gruenberger R, Jungwirth H, Fröhlich KU, Sinner F, Kroemer G, Madeo F. The propeptide of yeast cathepsin D inhibits programmed necrosis. Cell Death Dis 2011; 2: e161.
263. Choi S, Keys H, Staples RJ, Yuan J, Degterev A, Cuny GD. Optimization of tricyclic Nec-3 necroptosis inhibitors for in vitro liver microsomal stability. Bioorg Med Chem Lett 2012; 22: 5685-5688.
264. Zheng W, Degterev A, Hsu E, Yuan J, Yuan C. Structure-activity relationship study of a novel necroptosis inhibitor, necrostatin-7. Bioorg Med Chem Lett 2008; 18: 4932-4935.
265. Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, Levy JM, Pollyea DA, Jordan CT, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 2015; 25: 707-725.
266. Seneviratne D, Ma J, Tan X, Kwon YK, Muhammad E, Melhem M, DeFrances MC, Zarnegar R. Genomic instability causes HGF gene activation in colon cancer cells, promoting their resistance to necroptosis. Gastroenterology 2015; 148: 181-191.
267. Greijer AE, Delis-van Diemen PM, Fijneman RJ, Giles RH, Voest EE, van Hinsbergh VW, Meijer GA. Presence of HIF-1 and related genes in normal mucosa, adenomas and carcinomas of the colorectum. Virchows Arch 2008; 452: 535-544.
268. Airley R, Evans A, Mobasheri A, Hewitt SM. Glucose transporter Glut-1 is detectable in peri-necrotic regions in many human tumor types but not normal tissues: Study using tissue microarrays. Ann Anat 2010; 192: 133-138.
269. Huang CY, Kuo WT, Huang YC, Lee TC, Yu LC. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis 2013; 4: e622.