
INTRODUCTION
Synthetic lethality describes the genetic interaction by which the combination of two separately non-lethal mutations results in lethality. The phenomenon was first described by Calvin Bridges in 1922 [1], who noticed that some combinations of mutations in the model organism Drosophila melanogaster confer lethality. Generally, the ablation of two genes located in parallel pathways (leading to cell survival or a common essential product) is one of the important patterns causing synthetic lethality (SL) [2].
Cancer is fundamentally a genetic disease with numerous gene mutations involved. Some of these genetic mutations serve as biomarkers in cancers. In particular, notable advances have been made in cancer therapy for example, with the discovery of Herceptin to treat breast cancer patients with HER2 amplification, and with Iressa for the treatment of non-small cell lung cancer patients with an EGFR mutation. However, developing drugs that selectively kill cancer cells without harming normal cells remains a big challenge in oncology therapy. Given that genetic mutations underpin differences between cancer cells and healthy cells, Hartwell [3] was the first to suggest the use of chemical and genetic synthetic lethality screening for cancer therapy. Since then, this approach has attracted great attention from cancer biologists as it provides a promising perspective for oncology medicine discovery [4, 5]. For example, targeting the PARP-1 enzyme using Olaparib in ovarian cancer patients carrying a tumor BRCA1/2 mutation achieved milestone success in this area [6]. Ultimately, siRNA and CRISPR screenings are the most reliable methods for detecting SL gene pairs. However, compared to model genetic systems (such as yeast or fruit flies), human cell systems hold greater challenges for genome-wide siRNA or CRISPR screening. For this reason, several computational approaches have been proposed to facilitate the systematic detection of SL gene pairs in cancer. Briefly, these methods can be divided into three categories according to their targeted data resources: (i) inferring human ortholog gene pairs from yeast SL genes [7]; (ii) using the robustness features to evaluate the importance of gene pairs in the cancer PPI network [8]; (iii) calculating mutual exclusivity using statistical models from gene mutation/transcriptional expression data [9–12]. More recently, Livnat et al. [13] proposed DAISY to identify SL gene pairs. This approach combines somatic copy number alteration, siRNA screening as well as cell survival and gene co-expression information. Derived from comprehensive in-house data, this approach achieved a promising performance in data-driving SL gene pair identification. Nevertheless, we comprehensively compared the four available predicted SL data sets from previously developed methods (including DAISY) [8, 10, 12, 13] on SL gene pair prediction. The concordance of predicted SL gene pairs among those different methods is extremely low (see details in Discussions). This inconsistency across different methods may indicate that the in silico SL gene pair identification methods are far from mature. In addition, none of the previous methods was learning-based, that is, SL gene pair identification was based on the screening of certain criteria rather than training and prediction. We noticed that a portion of known SL gene pairs have been accumulated, and the investigation of the characteristics of these SL gene pairs are expected to derive significant features which can quantitatively depict the common mechanisms of the SL. Therefore, in our study, we designed a learning-based pipeline to rank novel SL gene pairs based on the known SL gene pairs, together with other unknown ones. By mining the accumulated TCGA mutation and gene expression data, as well as the gene properties in the protein-protein interaction network, our pipeline can be treated as an integration of the traditional strategies, and ranked a list of potential SL gene pairs. In contrast to the lack of experimental validation in most previous methods, we implemented further siRNA knock-down experiments to evaluate our results.
RESULTS
Brief results of 10 times 5-fold cross validation
We evaluated the ranking performance in 11 cancers through 10 times 5-fold cross validation, the other cancers in TCGA failed due to the limited number of overlapping samples between mutation data and the expression data or limited coverage of positive SL pairs. The brief results were listed in Table 1. Herein we didn’t intend to describe the details of each pair, Kidney renal clear cell carcinoma (KIRC) would be picked out as an example for illustration (see details in Supplementary Table S1). Currently, TCGA mutation data contains 417 KIRC patients. In addition, the gene-expression data on 400 KIRC patients is available in the TCGA dataset. At first, 528 genes with mutation rate >= 1% were selected from the gene mutation data. Following the workflow described in methods, 1014 candidate SL gene pairs were generated with the chi-square test p value <=0.05 and mutation exclusivity ≥0.8. Three features (Gene pair mutation coverage, Driver mutation probability, Network information centrality) were subsequently calculated. The same calculation process was used on the 119 positive SL gene pairs covered by KIRC mutation and expression data and the cancer network. During the 10 times 5-fold cross validation procedure, the test set contained 23 or 24 SL pairs. Then the top 25 results were used for NDCG calculation and enrichment evaluation, respectively. alpha optimization is a critical process in data manifolds ranking algorithm, which can directly influence the ranking performance. As it was shown in Figure 1, NDCG@25 was gradually increased from 0 to 0.9768, as the increase of alpha; while the enrichment p value was decreased. This means that the ranking performance was better at a larger alpha. Finally, the optimized alpha = 0.84 was achieved when NDCG@25 reached the peak value. Then, after all of the positive SL pairs were imported as the training set with the optimized alpha, we generated a ranking list for the 1014 candidate pairs according to their relevance to positive pairs. The same process was implemented on 10 other cancer types. Finally, we generated a SL network in Figure 2, which is comprised of 107 predicted SL gene pairs from the top 10 results in the 11 cancer types. (See all of the ranking results in Supplementary Table S2)
Table 1: Ranking performance in 11 cancer types
Cancer Type |
Candidate pair Num. |
Positive pair Num. |
NDCG@Positive pair/5 |
Enriched p value |
Optimized alpha |
|---|---|---|---|---|---|
LGG |
1192 |
76 |
0.9808 |
5.96E-13 |
0.56 |
KIRC |
1014 |
119 |
0.9768 |
3.90E-13 |
0.84 |
CESC |
93 |
38 |
0.9343 |
0.0027 |
0.98 |
OV |
126 |
99 |
0.5574 |
1.50E-07 |
0.86 |
BRCA |
534 |
148 |
0.4618 |
0.0099 |
0.99 |
GBM |
791 |
49 |
0.3417 |
0.0262 |
0.99 |
LUAD |
766 |
164 |
0.3244 |
0.0609 |
0.99 |
LUSC |
2177 |
101 |
0.2928 |
5.29E-03 |
0.99 |
SKCM |
7290 |
135 |
0.2707 |
6.24E-04 |
0.98 |
HNSC |
2213 |
140 |
0.2643 |
0.0271 |
0.87 |
STAD |
1417 |
135 |
0.2372 |
0.0160 |
0.98 |
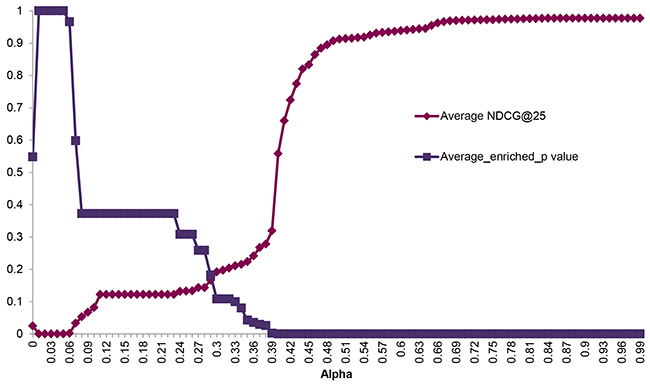
Figure 1: Ranking performance according to alpha. The X-axis indicates the parameter alpha in manifold ranking algorithm. The Y-axis represents the corresponding enriched p value and NDCG in the top 25 ranking results.
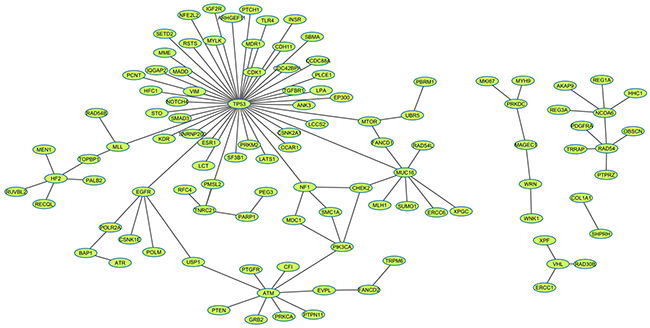
Figure 2: SL network of the predicted 107 pairs. Each node represents a gene. The SL relationship of a gene pair was linked by an edge. The graph was generated by Cytoscape.
Function analysis of the genes in the predicted novel SL pairs
Pathway enrichment was utilized to decipher the biological functions of the gene list. Specifically, 73/107 genes were mapped to KEGG pathways. From Table 2, we can see that these genes are mainly enriched in 15 pathways (covering 61.64% of the mapped genes) involved in six biological process categories, namely, replication and repair, signal transduction, the endocrine system, cell growth and death, cellular immunity and development. In particular, the Fanconi Anemia pathway ranked No.1 as a potential pathway for identifying novel anticancer therapies by exploiting synthetic lethal relationships [14]. The most famous example of this is the synthetic lethality relationship between the BRCA1/2 gene in the Fanconi Anemia pathways and PARP [15]. Recently, the first-in-class PARP inhibitor, Olaparib, was approved by the U.S. FDA for use in advanced ovarian cancer patients with BRCA mutations [16]. Based on our findings, several other biological pathways for synthetic lethality exploration were identified. For example, the HIF-1 signalling pathway (which activates the transcription of genes involved in angiogenesis, cell survival, glucose metabolism and invasion), was used as a screening resource in discovering synthetic lethal gene pairs [17]. RAS signalling [18], P53 signalling [19], PI3K-AKT signalling [20], are also widely considered to be promising pathways for synthetic lethal pair identification, and have previously attracted considerable research interest.
Table 2: Enriched pathways of the top 10 results in 11 cancer types
Pathway Category |
Pathway |
Predicted SL genes in the pathway |
Number of the predicted SL genes |
p value |
|---|---|---|---|---|
Replication and repair |
Fanconi anemia pathway |
ERCC1; PMS2; USP1; PALB2; ATR; FANCD2; BRCA2; ERCC4; MLH1; POLI |
10 |
3.06E-08 |
Signal transduction |
FoxO signaling pathway |
ATM; TGFBR1; PIK3CA; PTEN; INSR; CREBBP; EP300; SMAD3; GRB2; EGFR; MAPK1; CSNK1E |
12 |
7.02E-07 |
Signal transduction |
HIF-1 signaling pathway |
VHL; PIK3CA; INSR; CREBBP; MTOR; EP300; EGFR; TLR4; PRKCA; MAPK1 |
10 |
8.57E-06 |
Endocrine system |
Thyroid hormone signaling pathway |
TP53; PIK3CA; CREBBP; MTOR; EP300; NOTCH4; ESR1; PLCE1; PRKCA; MAPK1 |
10 |
1.55E-05 |
Cell growth and death |
Cell cycle |
TP53; ATM; CDK1; PRKDC; CREBBP; CHEK2; EP300; ATR; SMAD3; SMC1A |
10 |
2.68E-05 |
Cellular community |
Adherens junction |
TGFBR1; INSR; CREBBP; EP300; SMAD3; EGFR; CSNK2A1; MAPK1 |
8 |
3.60E-05 |
Signal transduction |
Ras signaling pathway |
PDGFRA; PIK3CA; INSR; PTPN11; GRB2; EGFR; NF1; KDR; PLCE1; PRKCA; MAPK1 |
11 |
0.00039 |
Cellular community |
Focal adhesion |
PDGFRA; PIK3CA; PTEN; COL1A1; MYLK; GRB2; EGFR; KDR; PRKCA; MAPK1 |
10 |
0.00067 |
Signal transduction |
ErbB signaling pathway |
PIK3CA; ERBB3; MTOR; GRB2; EGFR; PRKCA; MAPK1 |
7 |
0.00071 |
Signal transduction |
PI3K-AKT signaling pathway |
PDGFRA; TP53; PIK3CA; PTEN; COL1A1; INSR; MTOR; GRB2; EGFR; KDR; TLR4; PRKCA; MAPK1 |
13 |
0.00087 |
Cell growth and death |
p53 signaling pathway |
TP53; ATM; CDK1; PTEN; CHEK2; ATR |
6 |
0.0016 |
Replication and repair |
Nucleotide excision repair |
ERCC1; ERCC5; ERCC4; RFC4; ERCC6 |
5 |
0.0031 |
Development |
Dorso-ventral axis formation |
GRB2; EGFR; NOTCH4; MAPK1 |
4 |
0.0041 |
Cellular community |
Gap junction |
PDGFRA; CDK1; GRB2; EGFR; PRKCA; MAPK1 |
6 |
0.0049 |
Signal transduction |
Wnt signaling pathway |
TP53; CREBBP; EP300; SMAD3; CSNK2A1; PRKCA; CSNK1E |
7 |
0.0098 |
In-vitro drug sensitivity of cell lines in which one of the genes in each synthetic lethality pair is targeted
In the process of validating the SL gene pairs in the cell lines, two strict criteria are applied in the cell response information for each SL gene pair: 1. One of the genes in a SL pair should be the target of a drug in the database. 2. Several cancer cell lines treated by the drug must possess the mutation of the other gene in the SL pair. Finally, only 37 predicted SL gene pairs could be well annotated by the drug sensitivity data in CCLE [21] and NCI60 [22] databases (see Table 3 and Table 4). We compared the drug sensitivity of the two types of cancer cell lines, namely, the cell lines with the other gene mutation and the cell lines without the other gene mutation in the SL pair. We found 4 pairs: mTOR-TP53, VEGFR2-TP53, EGFR-TP53, ATM-PRKCA which showed significantly higher drug sensitivity (targeting one of the genes in the SL pairs) in the cell lines with the other gene mutations (P value <= 0.05), than the cell lines without the other gene mutation in these predicted SL pairs.
Table 3: Comparison of drug sensitivity between two groups of cell lines in CCLE data
Pair(Entrz Gene ID) |
Gene A |
Gene B |
Drug targeted on gene A |
Mean of drug sensitivity (PIC50)§ |
p value |
|
|---|---|---|---|---|---|---|
Cell lines with gene B mutation |
Cell lines with gene B wild type |
|||||
1009_7157 |
TP53 |
CDH11 |
Nutlin-3 |
5.0978 |
5.1246 |
0.7689 |
11200_4763 |
CHEK2 |
NF1 |
AZD7762 |
6.1902 |
6.0711 |
0.1553 |
11200_5290 |
PIK3CA |
CHEK2 |
GDC0941 |
5.1880 |
5.1031 |
0.3676 |
PIK3CA |
CHEK2 |
NVP-BEZ235 |
6.9181 |
6.9876 |
0.6393 |
|
CHEK2 |
PIK3CA |
AZD7762 |
5.8728 |
6.1251 |
0.9818 |
|
1387_7157 |
TP53 |
RSTS |
Nutlin-3 |
5.1032 |
5.1261 |
0.8182 |
1956_1454 |
EGFR |
CSNK1E |
Gefitinib |
5.1213 |
4.9910 |
0.2948 |
EGFR |
CSNK1E |
Lapatinib |
5.1555 |
5.0623 |
0.3428 |
|
EGFR |
CSNK1E |
BIBW2992 |
5.1208 |
5.0253 |
0.3775 |
|
EGFR |
CSNK1E |
Erlotinib |
5.0803 |
5.0415 |
0.4258 |
|
EGFR |
CSNK1E |
ZD-6474 |
5.1210 |
5.2915 |
0.8464 |
|
1956_7157 |
EGFR |
TP53 |
BIBW2992 |
5.1074 |
4.8862 |
0.0047 |
EGFR |
TP53 |
Gefitinib |
5.0485 |
4.8969 |
0.0121 |
|
EGFR |
TP53 |
ZD-6474 |
5.3078 |
5.2584 |
0.0679 |
|
EGFR |
TP53 |
Erlotinib |
5.0587 |
5.0137 |
0.1822 |
|
EGFR |
TP53 |
Lapatinib |
5.0810 |
5.0339 |
0.1933 |
|
TP53 |
EGFR |
Nutlin-3 |
5.1081 |
5.1247 |
0.7205 |
|
2065_7157 |
TP53 |
LCCS2 |
Nutlin-3 |
5.1057 |
5.1251 |
0.7572 |
2475_51366 |
MTOR |
UBR5 |
Temsirolimus |
6.4909 |
6.4765 |
0.4590 |
2475_7157 |
TP53 |
MTOR |
Nutlin-3 |
5.1135 |
5.1240 |
0.6411 |
MTOR |
TP53 |
Temsirolimus |
6.4561 |
6.5175 |
0.7357 |
|
3643_7157 |
TP53 |
INSR |
Nutlin-3 |
5.1204 |
5.1232 |
0.5272 |
367_7157 |
TP53 |
SBMA |
Nutlin-3 |
5.0970 |
5.1248 |
0.7850 |
3791_7157 |
TP53 |
VEGFR2 |
Nutlin-3 |
5.1186 |
5.1234 |
0.5590 |
VEGFR2 |
TP53 |
Sorafenib |
5.0474 |
5.1525 |
0.9738 |
|
4297_7157 |
TP53 |
MLL |
Nutlin-3 |
5.1239 |
5.1230 |
0.4885 |
4638_7157 |
TP53 |
MYLK |
Nutlin-3 |
5.1091 |
5.1320 |
0.9036 |
472_2885 |
ATM |
GRB2 |
KU-55933 |
3.6089 |
3.8219 |
0.8999 |
472_3426 |
ATM |
CFI |
KU-55933 |
3.7372 |
3.8174 |
0.6780 |
472_5290 |
PIK3CA |
ATM |
GDC0941 |
5.1010 |
5.1066 |
0.5171 |
ATM |
PIK3CA |
KU-55933 |
3.7713 |
3.8224 |
0.7299 |
|
PIK3CA |
ATM |
NVP-BEZ235 |
6.9180 |
6.9958 |
0.7795 |
|
472_5578 |
ATM |
PRKCA |
KU-55933 |
3.9089 |
3.7880 |
0.0423 |
472_5728 |
ATM |
PTEN |
KU-55933 |
3.8008 |
3.8179 |
0.5858 |
472_5781 |
ATM |
PTPN11 |
KU-55933 |
3.6390 |
3.8214 |
0.8728 |
4763_7157 |
TP53 |
NF1 |
Nutlin-3 |
5.1033 |
5.1260 |
0.8145 |
4855_7157 |
TP53 |
NOTCH4 |
Nutlin-3 |
5.1386 |
5.1184 |
0.1609 |
546_5156 |
PDGFRA |
RAD54 |
Pazopanib |
3.8402 |
4.1592 |
0.9844 |
PDGFRA |
RAD54 |
Sorafenib |
4.9588 |
5.1008 |
0.9495 |
|
5727_7157 |
TP53 |
PTCH1 |
Nutlin-3 |
5.1016 |
5.1248 |
0.7610 |
64324_7157 |
TP53 |
STO |
Nutlin-3 |
5.1031 |
5.1244 |
0.7307 |
2475_675 |
MTOR |
BRCA2 |
Temsirolimus |
6.3225 |
6.4942 |
0.8553 |
7099_7157 |
TP53 |
TLR4 |
Nutlin-3 |
5.1121 |
5.1236 |
0.6151 |
7157_1457 |
TP53 |
CSNK2A1 |
Nutlin-3 |
5.0969 |
5.1236 |
0.6783 |
7157_2033 |
TP53 |
EP300 |
Nutlin-3 |
5.1252 |
5.1219 |
0.4262 |
7157_2099 |
TP53 |
ESR1 |
Nutlin-3 |
5.0986 |
5.1238 |
0.6970 |
7157_4088 |
TP53 |
SMAD3 |
Nutlin-3 |
5.1190 |
5.1231 |
0.5239 |
7157_5594 |
TP53 |
PRKM2 |
Nutlin-3 |
5.0969 |
5.1233 |
0.6144 |
7157_7046 |
TP53 |
TGFBR1 |
Nutlin-3 |
5.0969 |
5.1240 |
0.7224 |
7157_983 |
TP53 |
CDK1 |
Nutlin-3 |
5.0969 |
5.1233 |
0.6276 |
CDK1 |
TP53 |
RO-3306 |
4.0215 |
4.0474 |
0.6447 |
|
8476_7157 |
TP53 |
CDC42BPA |
Nutlin-3 |
5.1285 |
5.1226 |
0.4244 |
9113_7157 |
TP53 |
LATS1 |
Nutlin-3 |
5.0969 |
5.1247 |
0.7767 |
§ PIC50 means negative log10(IC50) values (higher value indicate higher drug sensitivity)
Table 4: Comparison of drug sensitivity between two groups of cell lines in NCI60 data
Pair (Entrz Gene ID) |
GeneA |
Gene B |
Drug targeted on gene A |
Mean of drug sensitivity (z-score normalized GI50 values)* |
p value |
|
|---|---|---|---|---|---|---|
Cell lines with gene B mutation |
Cell lines with gene B wild type |
|||||
1956_7157 |
EGFR |
TP53 |
Lapatinib |
-0.1547 |
0.2300 |
0.0791 |
EGFR |
TP53 |
Erlotinib hydrochloride |
-0.0997 |
0.0925 |
0.2517 |
|
EGFR |
TP53 |
Gefitinib |
-0.0414 |
0.1119 |
0.2968 |
|
EGFR |
TP53 |
Afatinib |
-0.0551 |
-0.0219 |
0.4541 |
|
2475_7157 |
MTOR |
TP53 |
Sirolimus |
-0.2149 |
0.3950 |
0.0070 |
MTOR |
TP53 |
Temsirolimus |
-0.1846 |
0.4443 |
0.0146 |
|
MTOR |
TP53 |
Everolimus |
-0.1158 |
0.2619 |
0.0771 |
|
367_7157 |
SBMA |
TP53 |
Dromostanolone Propionate |
-0.1572 |
0.2575 |
0.0519 |
SBMA |
TP53 |
Calusterone |
-0.0156 |
0.1200 |
0.3373 |
|
SBMA |
TP53 |
Nandrolone phenpropionate |
0.0200 |
-0.0169 |
0.5508 |
|
3791_7157 |
VEGFR2 |
TP53 |
Pazopanib hydrochloride |
-0.2302 |
0.4594 |
0.0052 |
VEGFR2 |
TP53 |
Axitinib |
-0.1028 |
0.2731 |
0.1062 |
|
VEGFR2 |
TP53 |
Sunitinib malate/Sunitinib (free base) |
-0.0886 |
0.17125 |
0.1696 |
|
7157_2099 |
ESR1 |
TP53 |
Raloxifene hydrochloride |
-0.1808 |
0.2231 |
0.0740 |
ESR1 |
TP53 |
Fulvestrant |
-0.1194 |
0.2606 |
0.1093 |
|
ESR1 |
TP53 |
Tamoxifen citrate |
-0.0158 |
0.0525 |
0.3947 |
|
ESR1 |
TP53 |
Estramustine phosphate sodium |
-0.1011 |
-0.0450 |
0.4061 |
|
7157_5594 |
PRKM2 |
TP53 |
Arsenic Trioxide |
-0.0272 |
0.0275 |
0.4218 |
*z score normalized GI50 values are the elements of cellular fingerprint in NCI60 dataset. (http://data-analysis.charite.de/care/index.php?site=about#usecase) (Smaller values indicate higher drug sensitivity)
Possible molecular mechanisms of the 4 positive pairs
According to Kaelin [23]’s synthetic lethality model, synthetic lethality occurs via 4 different mechanisms: The cellular organizational units may be uniquely redundant and their roles are essential (type A), subunits of an essential multi-protein complex (type B), interconnected components in an essential linear pathway (type C), or they may participate in parallel pathways that are together essential (type D). The 4 pairs were consistent with either type D or type C.
mTOR-TP53
mTOR can integrate nutrient and mitogen signals to activate cell growth (increase cell mass and cell size) and cell division [24, 25], whilst one of the most important functions of TP53 is its ability to activate apoptosis [26]. Cell growth and apoptosis may provide parallel functions in cancer pathology. mTOR and TP53 may be considered synthetic lethality targets.
VEGFR2-TP53, EGFR-TP53
EGFR is a hot target for cancer therapy with many currently FDA approved drugs, and can activate at least 4 major downstream signalling cascades including; RAS-RAF-MEK-ERK, PI3 kinase-AKT, PLCgamma-PKC and STATs modules. Those signalling cascades can ultimately lead to a series of cellular events such as cell proliferation, inhibition of apoptosis, angiogenesis, migration, adhesion and invasion [27]. Furthermore, VEGF’s can specifically induce blood and lymphatic vessel development and homeostasis [28]. Inhibition of EGFR can block the angiogenesis process. Double knock out VEGFR2 and TP53 may lead to synthetic lethality through angiogenesis and apoptosis.
ATM-PRKCA
ATM [29] is a key regulator of multiple signaling cascades that respond to DNA damage. These responses involve the activation of cell cycle checkpoint factors, DNA repair and apoptosis. PRKCA has long been recognized to participate in activating tumour growth and development across different cancers [30]. In addition, PRKCA activation can result in increased cell motility in several in vivo and in vitro cancer models, the effect of which may be reversed with PRKCA inhibition [31, 32]. Hence, ATM and PRKCA knock-out, coupled with loss of function of apoptosis and the cell migration process, may generate synthetic lethality.
Validation through siRNA knock-down in cancer cell lines
Regarding the mutation information in the CancerDR database [33], three cancer cell lines were selected (Table 5). MCF-7 is a breast cancer cell line, carrying wild type TP53 and PRKCA. FaDu is a human epithelial cell line with mutant TP53 and SW48 is an invasive human colon adenocarcinoma cell line with a PRKCA mutation. Since an extremely low level of VEGFR2 mRNA expression was detected in the FaDu line, only three gene pairs (TP53-mTOR, TP53-EGFR, PRKCA-ATM) were able to be analyzed for siRNA knock-down validation. The relative cell growth results are displayed in Figure 3.
Table 5: siRNA knock-down on the cancer cell lines
SL pair (Entrz Gene ID) |
Gene A |
Cell line with wild type gene A |
Cell line with mutant gene A |
siRNA knock down gene B |
|---|---|---|---|---|
7157-2475 |
TP53 |
MCF-7 |
FaDu |
MTOR |
7157-1956 |
TP53 |
MCF-7 |
FaDu |
EGFR |
7157-3791 |
TP53 |
MCF-7 |
FaDu |
VEGFR2 |
472-5578 |
PRKCA |
MCF-7 |
SW48 |
ATM |
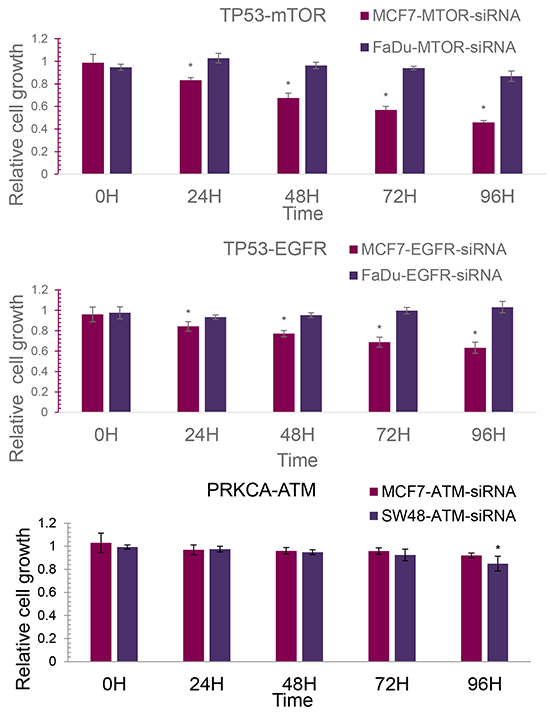
Figure 3: The relative cell growth of cancer cell lines. A. The relative cell growth of MCF7 and FaDu with siRNA knock-down of mTOR. B. The relative cell growth of MCF7 and FaDu with siRNA knock-down of EGFR. C. The relative cell growth of MCF7 and SW48 with siRNA knock-down of ATM.
siRNA knock-down validation on TP53-mTOR
According to the results of mRNA expression detection, there was no significant difference in the mTOR knock-down rate between MCF7 and FaDu (average mTOR knock-down rate: MCF7 62.26%; FaDu 60.67%). As shown in Figure 3A, the cell growth inhibitory effect of mTOR knock-down was observed in both MCF7 and FaDu. 96 hours after mTOR siRNA knock-down, the relative cell growth of FaDu slightly decreased from 1.1596 to 0.8178, while the relative cell growth of MCF7 dramatically decreased from 0.9509 to 0.4581. 24 hours after mTOR knock-down, the relative cell growth of MCF7 was always significantly lower than FaDu with a t-test P value of less than 0.01. The lowest inhibition of cell growth (54.19%) was achieved after 96 hours of mTOR knockdown in the MCF7 cells. This may indicate that wild type TP53 in the MCF7 line can possibly strongly enhance the cell growth inhibition effects of mTOR knock-down, compared to the mutant TP53 in FaDu.
siRNA knock-down validation on TP53-EGFR
Also, no significant difference in the EGFR siRNA knock-down rate was detected between MCF7 and FaDu. The optimal knock-down rates were 82.81% and 88.87% in FaDu and MCF7, respectively. Figure 3B displays the relative cell growth of MCF7 and FaDu cells after EGFR siRNA knock-down within 96 hours. No inhibition of cell growth using EGFR knock-down was observed in FaDu cells. The average relative cell growth was maintained at around 0.9325 ~ 1.0300 upon EGFR knock-down, whilst the relative cell growth of MCF7 strongly decreased from 0.9595 to 0.6884 following EGFR knock-down. 24 hours after EGFR knock-down, the relative cell growth of the MCF7 line was always significantly lower than the FaDu line with a t-test P value of less than 0.01. The wild type TP53 in MCF7 cells with EGFR knock-down could lead to cell growth inhibition.
siRNA knock-down validation on PRKCA-ATM
Upon siRNA transfection, the average knock-down rates of ATM were 86.07% and 45.43% in MCF7 and SW48 cells, respectively.Figure 3C shows the relative cell growth of MCF7 and SW48 cells with ATM siRNA knock-down. The relative cell growth of MCF cells decreased slightly from 1.030 to 0.9208. Considering the high ATM siRNA knock-down efficiency, this suggests that ATM knock-down has very limited inhibition effects on MCF cell growth. For SW48 cells, the relative cell growth decreased from 0.9936 to 0.8495. 96 hours after ATM knock-down, the relative cell growth of SW48 was significantly lower than MCF7 (0.8495 ± 0.0209 vs. 0.9208 ±0.0636, t test P value = 0.01296). This may partly indicate that the mutant PRKCA in SW48 cells can enhance the inhibition of cell growth on ATM knock-down, compared to the wild type PRKCA in MCF7. The slight inhibition observed on SW48 cell growth may be caused by the low ATM siRNA knock-down rate in the cell. It may be a novel SL gene pair with further rigorous validation.
DISCUSSION
Regarding the predicted SL gene pairs: mTOR-TP53 and EGFR-TP53, we found that mTOR knock-down or EGFR knock-down could cause much stronger cell growth inhibition in cell lines with wild type TP53, compared to mutant TP53. It seems that the relationships between mTOR-TP53 and EGFR-TP53 are exactly opposed to the concept of synthetic lethality. Indeed, the multifunctional nature of mutant TP53 needs to be better understood. A series of earlier studies [34–37] had suggested that mutant TP53 not only represents the equivalent of wild type TP53 functional loss, but also acquires new functions in driving cell migration, invasion and metastasis. The significant differences in cell growth between cancer cell lines with wild type TP53 and mutant TP53 could partly suggest that there is a special relationship between TP53-mTOR and TP53-EGFR. Notably, TP53 may be a promising biomarker in the development of cancer drugs targeting the mTOR and EGFR pathways in precision medicine.
siRNA knock-down is limited by both the expression level of the gene in the cell and the knock-down efficiency of the siRNA. In our study, we failed to validate VEGFR2-TP53 due to the low expression levels of VEGFR2 in the FaDu cell line. The low knock-down rate (45%) of ATM may weaken the inhibitory effects on cell growth. In response to these issues, the latest CRISPR technology [38, 39], which can provide considerable gene editing power, may provide a more reliable approach to further validate SL gene pairs.
Detecting SL gene pairs in humans is a challenging problem due to the highly evolved, complex and redundant signalling pathways within human cells. The influence of a loss of function caused by gene mutation can often be complemented by parallel pathway signalling. Various computational methods can provide potential SL gene pairs from different perspectives, such as the correlation of gene expression with mutation, robustness in the cancer network or gene co-expression in related biological processes. In this study, we compared the 107 predicted SL pairs with the results of four previous methods (see details in Supplementary Table S3). As shown in Figure 4, 12.15% (13 pairs) of our predicted results overlapped with Wang’s [10] or Kranthi’s [8] prediction. Importantly, TP53-mTOR and EGFR-TP53 pairs validated by drug sensitivity data were included in the overlapping pairs. This may suggest that overlapping predictions from different methods may provide more reliable results. Interestingly, we also found that no overlap occurred between Livnat’s [13] predictions or any of the other four methods. The original input data may be one of the important factors in influencing the final predictions. Kranthi’s method [8] started with the human protein-protein interaction database HPRD [40] as well as CancerGenes [22]. Wang’s prediction [10] was based on the profiling data of glioblastoma multiforme from TCGA as well as the p53 mutation information from the Trust Sanger Institute. Srihari [12] used the copy-number and gene-expression profiling data of four cancers (breast, prostate, ovarian and uterine) in TCGA as input data for their method. The different features of these input data across the methods may generate bias in SL gene pair predictions. Since there was no priori knowledge of cancer targets in the NCI-60 database, CancerGenes or Metacore were used to filter the input data in Livnat’s model [13], the potential SL gene pairs from the cancer lines may lead to distinct prediction results from others. In addition, due to the low concordance of results between different methods, further efforts to explore such complex SL relationships in a human system may be required.
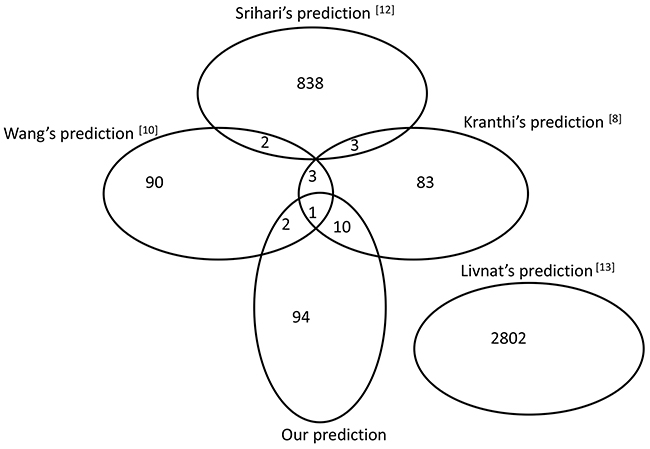
Figure 4: Comparison of the predicted results with other methods. The Venn diagram was drawn based on the overlap of the predicted SL gene pairs in three previous reports and our results.
CONCLUSIONS
In this study, we proposed a semi-supervised ranking pipeline to rank novel SL gene pairs based on the vast amounts of accumulated TCGA data. 107 novel potential SL gene pairs were predicted from the top 10 results covering 11 cancers. In particular, 4 SL pairs: mTOR-TP53, VEGFR2-TP53, EGFR-TP53, ATM-PRKCA, could be validated using drug sensitivity information in the cancer cell line databases CCLE or NCI60. Furthermore, the results of siRNA knock-down experiments indicated that significant differences in the cell growth of mTOR or EGFR siRNA knock-down were detected between the cancer cells with wild type TP53 and mutant TP53. The TP53 mutation may serve as a biomarker for cancer therapy in drugs targeting mTOR or EGFR. More promisingly, a recent study [41] has proposed P53 as a biomarker for predicting the progression free survival (PFS) of pancreatic cancer patients being treated with erlotinib (EGFR inhibitor). Taken together, these data underscore the potential of investigating the role of P53 as a predictive biomarker in other cancer types.
MATERIALS AND METHODS
SL gene pair prediction pipeline
In this study, we designed a semi-supervised learning model [42] to rank the similarities between positive SL gene pairs and candidate SL gene pairs, mainly using 3 defined features namely, gene pair mutation coverage, driver mutation probability and the quantified network information centrality. More specifically, we used three features to describe both the known SL gene pairs and candidate SL gene pairs. Then the semi-supervised method could rank the candidate SL gene pairs according to the similarity of these features with the known SL gene pairs. Herein, gene pair mutation coverage was defined as the percentage of samples containing at least one gene mutation in the pair. Furthermore, in order to get more reliable results from TCGA mutation data, the mutations of genes in candidate SL pairs should be covered by a certain number of samples. Driver mutations play vital roles in cancer development. Regarding the cancer specific SL pairs, we hypothesised that the mutation of genes playing an important role in cancer progression are more likely to be driver mutations. Last, the network information centrality helps to identify the potential nodes, which are crucial for the proper functioning of the system. Since simultaneously mutating two genes in a SL gene pair could dramatically influence the cellular process and cause cell death, network information centrality was used to calculate the influence of knocking-out a node pair on system stability. This approach inherently mimics the synthetic lethality mechanism well.
The brief workflow of the SL prediction pipeline is shown in Figure 5. In the first step, cancer biomarkers were collected from COSMIC [43] and MetaCore [44], which were used as a filter to select raw cancer related SL pairs. Next, the positive SL pairs were generated from yeast SL pairs, followed by homolog gene transformation, cancer biomarker filtering as well as the application of evidence in human cell lines obtained from literature mining. The candidate genes were selected from TCGA mutation data. The raw candidate SL pairs were then composed based on a candidate gene and a gene within a cancer network. Then, a Chi-square test (implemented by chi2_contingency in python package Scipy) was used to evaluate whether the mutations of the two genes is an independent event in each raw candidate SL pair. In addition, the mutation exclusivity was also calculated, which was defined as the percentage of samples carrying one of the mutant genes in the SL gene pair [9]. Only those independent gene mutations with high mutation exclusivity were selected as candidate SL pairs for further calculation. Subsequently, three features of both candidate SL pairs and positive SL pairs were calculated and normalized before being exported into a learning model. Finally, the novel SL pairs were detected with an optimized parameter which was obtained from 10 times 5-fold cross validation.
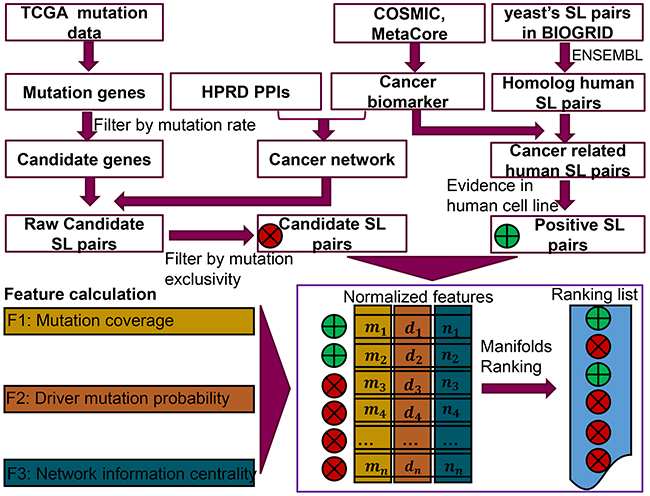
Figure 5: Workflow of the SL prediction pipeline. The three types of original data (gene mutation and expression, cancer biomarker, SL-pairs in yeast) were downloaded from TCGA, COSMIC and MetaCore as well as BIOGRID, respectively. The cancer network was built from the interaction of cancer biomarkers in the protein-protein interaction database HPRD. Each raw candidate SL pair was composed of a highly mutated gene and another gene in the cancer network. Then a chi-square test on the mutation exclusivity with p value <= 0.05 was utilized to generate the candidate SL pairs, while the positive SL pairs were derived from the yeasts’ SL pairs followed by homology transformation, cancer biomarker filtration and literature evidence identification on human cell lines. For each pair in of candidate SL and positive SL data, three features were generated for them. The first feature was calculated from the mutation coverage of each SL gene pair in the TCGA mutation data. The driver mutation probability was calculated by R package DriverNet. The third feature was defined to evaluate the influence on stability of the cancer network, after removing the two genes from an SL pair. Then normalized features of each SL pair were imported into a manifolds ranking model to generate a ranking list of potential SL pairs.
TCGA mutation and expression data processing
We downloaded TCGA mutation and expression profiling data from the UCSC Cancer Genomics Browser (https://genome-cancer.ucsc.edu), which provides well-annotated and interactive visualizations of TCGA genomic, phenotypic, and clinical data [45]. We then obtained two matrices. Each row of the matrices represented a gene, and each column indicated a sample. The values in the cells represented the expression and mutation status in the gene expression matrix and the gene mutation matrix, respectively. Finally, data from 11 cancers, containing both the gene expression matrix and the corresponding gene mutation matrix, were used in our study.
Positive synthetic lethality gene pairs
The collective data on yeast SL (synthetic lethal) genes based on high throughput genetic screening is available at BioGRID [46]. However, no curated database of human SL gene pairs has been established yet. In this study, BioGRID was used as the primary resource to retrieve human cancer related SL gene pairs. The phylogenetic inference from yeast to human genes was obtained from the Ensemble database (http://useast.ensembl.org/). Then, homolog human SL pairs were filtered by cancer biomarkers in MetaCore (https://portal.genego.com/) and driver genes in COSMIC [43]. Only homolog human SL gene pairs with both of the genes covered by cancer biomarkers or driver genes were kept for downstream analysis. In order to reduce the false positive rate as much as possible, for each homolog human SL gene pair, we checked the evidence available in the PubMed literature. Finally, 399 positive SL pairs were identified with the evidence of synthetic lethality in human cell lines or animal models in the literature (see Supplementary Table S4).
Cancer network
307,066 protein-protein interactions were downloaded from HPRD [40]. Then, we used cancer biomarkers from MetaCore and COSMIC [43] to filter them. In details, we searched the keywords ‘cancer, tumor, carcinoma’ in MetaCore and retrieved 4,296 cancer related biomarkers. At the same time, we also downloaded the 507 driver mutation genes collected in the Cancer Gene Census from the website of COSMIC. All of these gene mutations in Cancer Gene Census have been proved to causally implicate in cancer. Then, for each protein-protein interaction, only if both proteins are included in MetaCore cancer biomarkers or Cancer Gene Census in COSMIC, would the protein-protein interaction be kept. Finally, we obtained 11,925 protein-protein interaction pairs, corresponding to 2,869 individual proteins. The cancer network could be built with edge presented by the protein-protein interaction, as well as the node displayed by a protein.
Candidate SL pairs generation
We calculated the mutation rate of each gene among the samples in the TCGA mutation data. Herein, 1% was utilized as the cut-off threshold to select the candidate genes. Each raw candidate SL gene pair was generated by selecting a candidate gene as well as the other gene from the cancer network. Subsequently, we tested whether gene A mutation and gene B mutation are independent events based on the mutation data. In detail, the null hypothesis is that gene A mutation and gene B mutation are independent of each other. A Chi-square test was implemented on a 2×2 contingency table (see Table 6). M represents the number of samples carrying both gene A and gene B mutations; N represents the number of samples carrying the gene A mutation without the gene B mutation; X represents the number of samples carrying the gene B mutation without the gene A mutation; Y is the number of samples that containing both wild type gene A and wild type gene B.
Table 6: 2×2 contingency table in chi-square test
gene B + |
gene B − |
|
|---|---|---|
gene A + |
M |
N |
gene A − |
X |
Y |
+ mutant type
− wild type
The raw candidate SL gene pairs with Chi-square test p value <=0.05 means the mutation of gene A and gene B are not independent. Maybe some relationships exist between mutation of gene A and gene B. In addition, the mutation exclusivity of gene A and gene B could be calculated as (X+N)/(M+N+X). The higher mutation exclusivity indicates the gene A and gene B are more likely to be mutually exclusive mutations. Herein, only candidate SL pairs with both Chi-square test P value ≤ 0.05 and mutation exclusivity ≥ 0.8 were selected for the downstream processing.
Features calculation
Gene pair mutation coverage
It was defined as the percentage of samples containing at least one gene mutation in the pair. For example, gene A is mutated in samples s1,s3,s6, gene B is mutated in samples s3,s8,s9. n is the total number of samples. The mutation coverage of the pair (gene A, gene B) is 5/n.
Driver mutation probability
Herein, we utilized the R package DriverNet [47] to evaluate the driver mutation probability of genes based on the relationship between mutation and consequent changes in gene expression. The input data of DriverNet comes from two matrices, namely a mutation matrix and its corresponding gene expression matrix. Each column of the two matrices is a sample, whilst each row represents the mutation status or expression level of a gene among the samples. The output of DriverNet is the P value of each gene that will likely be a driver of gene mutation. The smaller P value of the two genes from a SL pair was transformed to a negative log10 (P value) indicating the strength of the driver mutation for the pair.
Network information centrality
If G refers to the cancer network mentioned above, and G’ refers to the cancer network after removing gene A and gene B, then the network information centrality of gene A and gene B could be defined as formula I:
Where E(G) is the efficiency of the network. It could be calculated in the formula II:
Herein, if gene i could reach gene j in cancer network, dij is the length of the shortest path between the gene i and gene j (calculated by shortest_path_length in python package networkx), otherwise, dij is equal to D(G) + 1. D(G) represents the diameter of cancer network, which is defined as the largest distance across all of the shortest paths in the cancer network (calculated through diameter in python package networkx).
Finally, normalization of the three features was taken to transform the values of each feature between 0-1 in formula III. x is the original value of a feature. x’ is the normalized value.
A semi-supervised ranking model
The principle of our ranking model, which is referred to as a manifold ranking algorithm [42, 48] can be intuitively explained: the problem is defined in two datasets, a true sample set and an unknown sample set (background); and the goal is to rank the individual members of the unknown sample set according to their relevance to the true samples. This model is well suited to address our problem scenario, which is that we only have few known SL pairs in hand (known positive data samples), and we want to prioritize the largest possible gene pair combinations based on their possibility to be the true SL pairs. In detail, we used three features to describe each SL pair. Then 1- cosine angle distance was calculated to represent the relevance between candidate SL pairs and true SL pairs.
Input: A set of points X = (x1... xq, xq + 1... xn) representing the SL pairs. The first q points are true SL pairs, while, the others are candidate SL pairs. The initial score y was defined as (1…1,0 …0). (The true SL pairs are corresponding to 1, candidate SL pairs are assigned as 0.) Define f 0 = y; α is a parameter of the algorithm.
Output: A ranked list of X, where higher ranked gene pairs are more likely to be SL gene pairs.
1. Define the similarity matrix Wij = 1- cosine(i,j) and Wii = 0.
2. Compute L = D-1/2WD-1/2 with D being a diagonal matrix defined as
3. Set iteratively f t+1 = αLf t + (1-α)y until f converges, where α is a parameter in [0, 1);
4. Let f * be the converged function f t; and rank all the points X in the decreasing order of their f * values.
It has been shown [42] that f * could be calculated as formula IV.
Evaluation test design
10 times 5-fold cross validation
For each case, the positive SL pairs were divided into five segments. Four of them were used as training sets, while the rest of the segments were used for evaluation. Next, positive SL pairs were shuffled 10 times, the overall performance was determined by the average results of these 10 shuffling events.
Ranking performance evaluation
Normalized discounted cumulative gain (NDCG) [49] was originally used to evaluate web search engine algorithms in the field of information retrieval. It can measure the usefulness of a document based on its position in the result list. Here we used NDCG to measure the effectiveness of ranking performance for each case’s predicted results (see formula V ).
Z is the normalization constant.
i is the rank position of candidate SL pair m.
reli is the relevance value of candidate SL pair m. If candidate SL pair m belongs to the positive SL pairs, reli is set to 1, otherwise, reli is set to 0.
p is the maximum position.
For example, if the three positive SL pairs are ranked at 2, 3 & 8, respectively, while the ideal rank position should be 1, 2 & 3, then:
In the top 5 results,
In addition, the positive enrichment of SL pairs in the top n ranking position are also used to evaluate our prediction performance. Herein, a hypergeometric test is utilized. (see formula VI)
k: number of positive SL pairs included in top n ranking results.
N: the whole candidate SL pairs
M: the whole positive SL pairs.
Comparison of drug sensitivities between two groups of cancer cell lines
The original drug sensitivity data, drug targets as well as the mutation backgrounds of cancer cell lines in CCLE [21], NCI60 [22] were downloaded from broadinstitute.org/ccle/home and discover.nci.nih.gov/cellminer/, respectively. Regarding a SL gene pair, if one of the genes in the SL pair is targeted by a drug, we compared the drug sensitivities on the cell lines carrying the mutation of the other genes in the pair and the cell lines containing the wild type of the other genes. The lower GI50 or IC50 value means higher drug sensitivity.
Further validation through siRNA knock down on cell lines
In order to get more reliable validation of the predicted SL gene pairs, we conducted siRNA knock-down experiments on cancer cell lines. The influence on cell growth of different genetic background cell lines would indicate SL relationships.
For example, regarding a SL gene pair gene a –gene b, two cancer cell lines were selected. The first cell line carried mutant gene a, while the wild type gene a was carried in the other cell line. Then, the siRNA of gene b was transfected into the two cell lines. We recorded the cell growth of two cell lines at the time points of 0h, 24h, 48h, 72h and 96h on two different treatments: placebo, siRNA-knockdown of gene b, respectively. Herein, we did 8 parallel experiments at each time point. The relative cell growth was calculated through the formula VII.
ACKNOWLEDGMENTS
Funding: National Natural Science Foundation of China (61572361, 81000438), Shanghai Rising-Star Program (Grant No. 16QA1403900) and Fundamental Research Funds for the Central Universities (2000219122, 1054209038, 1501219106).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011; 585:1-6.
2. Canaani D. Application of the concept synthetic lethality toward anticancer therapy: A promise fulfilled? Cancer letters. 2014; 352:59-65.
3. Hartwell LH, Szankasi P, Roberts CJ, Murray AW and Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997; 278:1064-1068.
4. Kaelin WG, Jr. Synthetic lethality: a framework for the development of wiser cancer therapeutics. Genome Med. 2009; 1:99.
5. Luo J, Solimini NL and Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009; 136:823-837.
6. Eskander RN and Tewari KS. PARP inhibition and synthetic lethality in ovarian cancer. Expert Rev Clin Pharmacol. 2014; 7:613-622.
7. Conde-Pueyo N, Munteanu A, Sole RV and Rodriguez-Caso C. Human synthetic lethal inference as potential anti-cancer target gene detection. BMC Syst Biol. 2009; 3:116.
8. Kranthi T, Rao SB and Manimaran P. Identification of synthetic lethal pairs in biological systems through network information centrality. Mol Biosyst. 2013; 9:2163-2167.
9. Miller CA, Settle SH, Sulman EP, Aldape KD and Milosavljevic A. Discovering functional modules by identifying recurrent and mutually exclusive mutational patterns in tumors. BMC Med Genomics. 2011; 4:34.
10. Wang X and Simon R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med Genomics. 2013; 6:30.
11. Ciriello G, Cerami E, Sander C and Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012; 22:398-406.
12. Srihari S, Singla J, Wong L and Ragan MA. Inferring synthetic lethal interactions from mutual exclusivity of genetic events in cancer. Biology direct. 2015; 10:57.
13. Jerby-Arnon L, Pfetzer N, Waldman YY, McGarry L, James D, Shanks E, Seashore-Ludlow B, Weinstock A, Geiger T and Clemons PA. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell. 2014; 158:1199-1209.
14. Jenkins C, Kan J and Hoatlin ME. Targeting the Fanconi Anemia Pathway to Identify Tailored Anticancer Therapeutics. Anemia. 2012; 2012:7.
15. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A and O’Connor MJ. Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. New England Journal of Medicine. 2009; 361:123-134.
16. Ray T. (2014). FDA Approves First PARP Inhibitor with Myriad’s BRACAnalysis as CDx in Advanced Ovarian Cancer. genomeweb: genomeweb).
17. Jones DT and Harris AL. Small-molecule inhibitors of the HIF pathway and synthetic lethal interactions. Expert Opinion on Therapeutic Targets. 2012; 16:463-480.
18. Weidle UH, Maisel D and Eick D. Synthetic Lethality-based Targets for Discovery of New Cancer Therapeutics. Cancer Genomics - Proteomics. 2011; 8:159-171.
19. Morandell S and Yaffe MB. Exploiting synthetic lethal interactions between DNA damage signaling, checkpoint control, and p53 for targeted cancer therapy. Prog Mol Biol Transl Sci. 2012; 110:289-314.
20. Crowder RJ, Phommaly C, Tao Y, Hoog J, Luo J, Perou CM, Parker JS, Miller MA, Huntsman DG and Lin L. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor–positive breast cancer. Cancer research. 2009; 69:3955-3962.
21. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV and Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603-607.
22. Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J and Pommier Y. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer research. 2012; 72:3499-3511.
23. Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nature reviews cancer. 2005; 5:689-698.
24. Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C and Blenis J. mTOR Controls Cell Cycle Progression through Its Cell Growth Effectors S6K1 and 4E-BP1/Eukaryotic Translation Initiation Factor 4E. Molecular and Cellular Biology. 2004; 24:200-216.
25. Laplante M and Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012; 149:274-293.
26. Fridman JS and Lowe SW. Control of apoptosis by p53. Oncogene. 2003; 22:9030-9040.
27. Oda K, Matsuoka Y, Funahashi A and Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Molecular systems biology. 2005; 1:2005 0010.
28. Cebe-Suarez S, Zehnder-Fjällman A and Ballmer-Hofer K. The role of VEGF receptors in angiogenesis; complex partnerships. Cellular and molecular life sciences. 2006; 63:601-615.
29. Lavin MF and Kozlov S. ATM activation and DNA damage response. Cell cycle. 2007; 6:931-942.
30. Kang J-H. Protein Kinase C (PKC) Isozymes and Cancer. New Journal of Science. 2014; 2014.
31. Koivunen J, Aaltonen V, Koskela S, Lehenkari P, Laato M and Peltonen J. Protein kinase C α/β inhibitor Go6976 promotes formation of cell junctions and inhibits invasion of urinary bladder carcinoma cells. Cancer research. 2004; 64:5693-5701.
32. Masur K, Lang K, Niggemann B, Zanker KS and Entschladen F. High PKC α and low E-cadherin expression contribute to high migratory activity of colon carcinoma cells. Molecular biology of the cell. 2001; 12:1973-1982.
33. Kumar R, Chaudhary K, Gupta S, Singh H, Kumar S, Gautam A, Kapoor P and Raghava GP. CancerDR: cancer drug resistance database. Scientific reports. 2013; 3:1445.
34. Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, Skinner HD, Fitzgerald AL, Osman AA, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Molecular cell. 2014; 54:960-974.
35. Oren M and Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harbor perspectives in biology. 2010; 2:a001107.
36. Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C and Levine AJ. Gain of function mutations in p53. Nature genetics. 1993; 4:42-46.
37. Muller PA and Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer cell. 2014; 25:304-317.
38. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W and Marraffini LA. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819-823.
39. Doudna JA and Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346:1258096.
40. Prasad TK, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B and Venugopal A. Human protein reference database—2009 update. Nucleic acids research. 2009; 37:D767-D772.
41. Ormanns S, Siveke JT, Heinemann V, Haas M, Sipos B, Schlitter AM, Esposito I, Jung A, Laubender RP, Kruger S, Vehling-Kaiser U, Winkelmann C, Fischer von Weikersthal L, et al. pERK, pAKT and p53 as tissue biomarkers in erlotinib-treated patients with advanced pancreatic cancer: a translational subgroup analysis from AIO-PK0104. BMC Cancer. 2014; 14:624.
42. Zhou D, Weston J, Gretton A and Bousquet O. Ranking on data manifolds. 2004.
43. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR and Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research. 2011; 39:D945-950.
44. Thomson Reuters MetaCore database: https://portal.genego.com/ (license from AstraZeneca).
45. Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D and Zhu J. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Scientific reports. 2013; 3:2652.
46. Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, Nixon J, Ramage L, Kolas N, O’Donnell L, Reguly T, Breitkreutz A, Sellam A, et al. The BioGRID interaction database: 2013 update. Nucleic acids research. 2013; 41:D816-823.
47. Bashashati A, Haffari G, Ding J, Ha G, Lui K, Rosner J, Huntsman DG, Caldas C, Aparicio SA and Shah SP. DriverNet: uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012; 13:R124.
48. Liu Q, Cui J, Yang Q and Xu Y. In-silico prediction of blood-secretory human proteins using a ranking algorithm. BMC bioinformatics. 2010; 11:250.
49. Järvelin K and Kekäläinen J. Cumulated gain-based evaluation of IR techniques. ACM Transactions on Information Systems (TOIS). 2002; 20:422-446.