
INTRODUCTION
Congenital hypothyroidism (CH) is one of the most common neonatal endocrine disorders, presenting with abnormal growth and intellectual impairment. Symptoms are caused by reduced thyroid function. CH occurs in 1 in 3000–4000 births and affects twice as many females as males [1, 2]. Without prompt thyroid hormone replacement, the physical and mental disability can become permanent. Clinical manifestations of CH mainly include poor feeding, prolonged jaundice, edematous, umbilical hernia, and dry skin. The majority of cases are sporadic, while hereditary cases show classical Mendelian autosomal recessive inheritance [3]. CH patients can be divided into two groups: about 85% of cases are caused by thyroid dysgenesis, including agenesis (22–42%), ectopy (35–42%), and hypoplasia (24–36%) [4], and the remaining 15% are associated with dyshormonogenesis. To date, a variety of gene mutations have been associated with thyroid hormone synthesis, such as thyroglobulin (TG) [5], thyroperoxidase (TPO) [6], sodium/iodide symporter (NIS) [7], dual oxidase 2 (DUOX2) [8], DUOX maturation factor 2 (DUOXA2) [9], pendrin (PDS) [10], and iodotyrosine deiodinase (DEHAL1) [11].
A central part of thyroid hormone synthesis is iodide organification, which requires thyroid peroxidase (TPO) and hydrogen peroxide (H2O2). H2O2 is used as a substrate in the iodination of tyrosine residues and the coupling of iodotyrosine residues to TG [12]. The DUOX1, DUOX2, DUOXA1 and DUOXA2 proteins have recently been identified as components of the H2O2 generation system in the thyroid. Among them, DUOX1 and DUOX2 are essential enzymes in the production of H2O2. DUOXA1 and DUOXA2 enable the DUOX proteins to translocate to the plasma membrane and become fully active [13]. DUOX1 and DUOX2 encode two very similar proteins that are inserted into the apical membrane of the thyroid follicular cell. DUOX2 expression is at least five times higher than DUOX1 [14, 15]. Defective H2O2 production causes CH. Defects in DUOX2 cause severe, permanent CH due to complete disruption of thyroid hormone synthesis or milder, transient hypothyroidism caused by insufficient quantities of thyroid hormones [13].
DUOX2 is located on chromosome 15q15.3 and consists of 34 exons, which of its 33 exons encode a 6376 nucleotide mRNA [16, 17]. The DUOX2 protein, a 1548-amino-acid polypeptide including a 26-amino-acid signal peptide, localizes to the apical membrane of thyrocytes and is involved in the Ca2+/reduced nicotinamide adenine dinucleotide phosphate (NADPH)-dependent generation of H2O2 [18]. The structure of these proteins includes seven putative transmembrane domains, four NADPH-binding sites, one flavin adenine dinucleotide (FAD)-binding site, and two EF-hand motifs that putatively control enzymatic activity through calcium binding [19].
The aim of this study was to screen DUOX2 mutations in 10 unrelated Chinese children with CH and goiter by standard polymerase chain reaction (PCR)-based sequencing and clarify genotype–phenotype relationships. We also examined the functional effect of the identified mutations on DUOX2 at the molecular level.
RESULTS
Screening of mutations
Direct sequencing of DUOX2 from 10 unrelated patients revealed two heterozygous missense mutations in two patients. One was a novel missense mutation (c.1060C>T), which results in an arginine to tryptophan substitution at codon 354 in exon 10 (p.R354W, Figure 1A). The other was a known missense variation (c.3616G>A), which predicts an alanine to threonine substitution at codon 1206 in exon 28 (p.A1206T, Figure 1B). However, no mutation was found in the remaining eight patients. Deleterious effects were predicted by Polymorphism Phenotyping v2 software (Polyphen) and Sorting intolerant from tolerant (SIFT) software. Polyphen predicts the possible impact of an amino acid substitution on the structure and function of a human protein using physical considerations and SIFT predicts whether an amino acid substitution affects protein function. Damaging effects on the DUOX2 protein were confirmed for both DUOX2 mutation as follows: p.R354W: SIFT score 0.01, PolyPhen-2 score 0.98; p.A1206T: SIFT score 0, PolyPhen-2 score 1. We did not find these two variants in 100 Chinese healthy controls. Mutation segregation with the phenotype within the family was not performed because data was lacking.
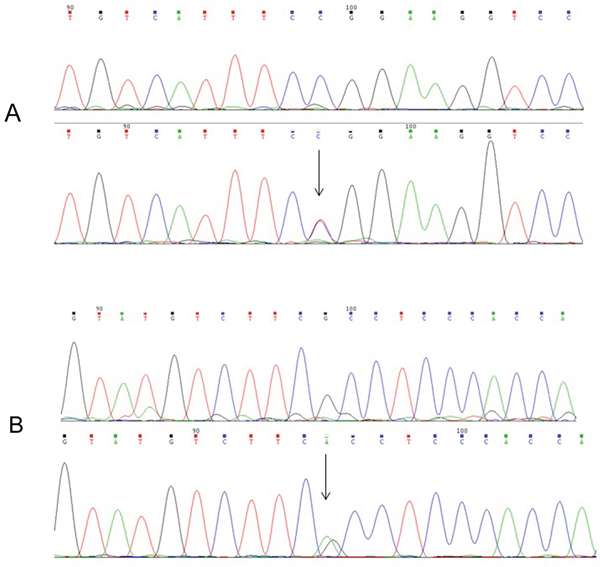
Figure 1: Partial sequence of exon 10 and exon 28 in the DUOX2 from WT(top) and mutational individuals (below). A. Arrowhead indicates the heterozygous C and T at nucleotide 1060 in an affected patient; B. Arrowhead indicates the heterozygous G and A at nucleotide 3616 in an affected patient.
Clinical data
Patient 1 with a p.R354W mutation was a male infant. He was born after full-term gestation by cesarean delivery from unrelated parents. His birth weight was 2,800 g. There was no family history of thyroid disease. Neonatal screening showed a high level of TSH (187.6 μIU/ml; upper limit, 20 μIU/ml), so he was recalled at the age of 23 days for further evaluation. At that time, his body weight was 3,600 g and his height/length was 53 cm. Biochemical evaluation showed a serum TSH level of 218 uIU/ml and a FT4 level of 4.2 pmol/L. A 99mTc thyroid scan confirmed goiter (Left lobe:12×10mm, Right lobe 13×10mm) and the perchlorate discharge test was positive (iodine uptake rate >30.51%). Levothyroxine (L-T4) replacement therapy was started immediately at an initial dose of 8.3 μg/kg per day. Replacement therapy was modified during follow-up according to clinical and hormonal evaluations to maintain normal serum TSH and FT4 levels. At 2.5 years of age, TSH, FT4, and FT3 concentrations were normal following a temporary withdrawal of L-T4 therapy for 4 weeks. Therefore, he was diagnosed with transient CH. The patient is now 4.5 years old. His physical and mental development is normal.
Patient 2 (P2), was a female subject with a p.A1206T mutation, born by cesarean delivery after full-term gestation to unrelated parents. Her birth weight was 2,650 g. There were no abnormal physical findings until neonatal screening at the age of 6 days, which revealed a high level of TSH (141.5 μIU/ml). The patient was diagnosed with CH according to the neonatal screening program. At the age of 21 days, the patient was recalled to confirm the test results, which showed a high TSH level (186.3 μIU/ml) and a low FT4 level (5.8 pmol/L). At the same time, a 99mTc thyroid scan showed a normally shaped orthotopic but mildly enlarged thyroid gland. L-T4 replacement therapy was started immediately at an initial dose of 8.8 ug/kg per day. The dose was adjusted according to clinical and hormonal evaluations to maintain normal serum TSH, FT4, and FT3 levels. When she was 2 years old, concentrations of TSH, FT4, and FT3 were normal following a temporary withdrawal of L-T4 therapy for 4 weeks. The patient is now 6 years old and her physical and mental development is normal, with no evidence of goiter.
H2O2 production assay
H2O2 generation was measured after either DUOX2 alone or both DUOX2 and DUOXA2 were expressed in 293T cells (Figure 2). Transfection of DUOX2 alone did not increase H2O2 production compared with nontransfected cells. The co-transfection of WT DUOX2 with DUOXA2 permits to DUOX2 to be active as indicated by the significant amounts of H2O2 released from the cells. Conversely, co-transfection of p.R354W and p.A1206T DUOX2 even in the presence of DUOXA2 did not show any activity, suggesting that these mutants were able to impair the functional activity of the H2O2-generating system.
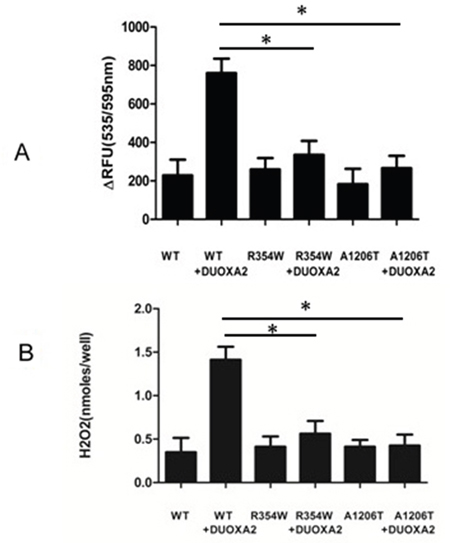
Figure 2: Showed H2O2 generation of cells transfected with the indicated expression vectors. The total amount of plasmids pertransfection was kept constant by adjusting with empty vector. A. RFU shown the changes in resorufin fluorescence vs. the baseline of empty vector transfected cells. B. Nanomoles of H2O2 was converted from fluorescence intensity using a calibration curve. (n=3, *p<0.05).
CHX chase experiments
Twenty-four hours after transfection with the indicated expression vectors, protein expression was inhibited by addition of CHX to the medium. Cell lysates were prepared at the indicated times thereafter, and equivalent amounts of total protein were immunoblotted. Compared with the WT DUOX2 proteins at the beginning of the incubation period (0 hours), DUOX2 mutants had shorter half-lives and degraded more rapidly (Figure 3A). One hour and 3 hours after transfection with the indicated expression vectors, there was no significant difference in the degradation of DUOX2 proteins. However, 15 hours and 24 hours after transfection, the degradation of both DUOX2 mutants was higher compared with the WT group (Figure 3B).
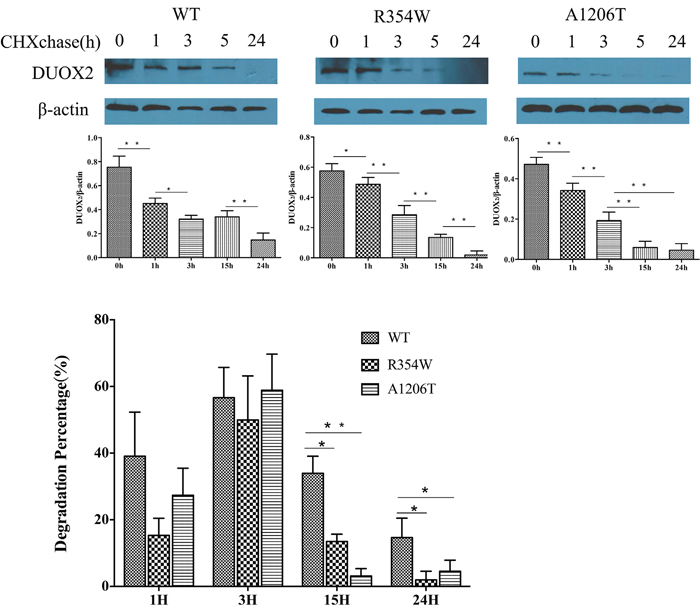
Figure 3: Cycloheximide(CHX) chase experiments. 100 μg/mL CHX was added to the medium of Hela transfected with DUOXA2 and either WT or one of the mutations DUOX2 expression plasmids. Western blot analysis showed the expression of DUOX2 (175kda) and β-actin(43kda). The expression was quantified densitometrically as the ratio of DUOX2/β-actin.(n=3, *p<0.05,**p<0.01).
DISCUSSION
The generation of hydrogen peroxide is a critical step in the synthesis of thyroid hormones. Mutation of DUOX2 causes hypothyroidism because of insufficient H2O2 production. DUOX proteins require DUOX maturation factors (DUOXA1 or DUOXA2) for the proper translocation of DUOX from the endoplasmic reticulum to the apical plasma membrane, where H2O2 production takes place. Since Moreno et al. reported the first DUOX2 mutation in 2002 [20], at least 35 more DUOX2 mutations have been identified, including missense mutations, nonsense mutations, frame shifts, and splice-site mutations [21, 22].
Grasberger used a DUOXA2 reconstituted system to characterize natural DUOX2 missense variants (p.Q36H, p.R376W, p.D506N) at the molecular level and analyze their impacts on H2O2 generation, trafficking, stability, folding and DUOXA2 interactions [23]. The p.Q36H and p. R376W mutations completely prevented translocation of DUOX2 to the cell surface. The p.D506N mutant displayed a partial deficiency phenotype with reduced surface expression but normal intrinsic H2O2-producing activity. The p.R376W [24] and p.Q36H [25] mutations were reported in permanent CH patients, while p.D506N [26] was found in temporary CH patients. Different DUOX2 mutations cause completely different CH phenotypes. In vitro experiments have revealed obvious pathogenic functions for some and none for others.
In our study, we described two missense mutations—p.R354W and p.A1206T in DUOX2. The novel mutation p.R354W is located in the extracellular peroxidase-like domain, and the known p.A1206T mutation is located in the 5th transmembrane helix segments. p.R354 and p.A1206 are highly conserved in other mammalian species and p.R354W and p.A1206T were not identified in 200 alleles from healthy subjects. The H2O2 production assay showed no impairment in H2O2-generating activity by WT DUOX2 protein or mutant proteins. Coexpression of WT proteins with DUOXA2 in HeLa cells significantly reconstituted H2O2 production. Mutant proteins were still unable to produce H2O2, which corresponded with the iodine organification defect observed in patients. We also found that mutant DUOX2 degraded faster than mature WT DUOX2.
Each patient had an increased TSH and decreased T4 serum level after birth. The only clinical sign of hypothyroidism was goiter. Euthyrox treatment was started immediately after the diagnosis of CH, and was continued until 2 years of age. When serum FT3 and FT4 levels had returned to normal, the growth and development were normal. This demonstrated that the phenotypes of the two affected patients were both transient and mild and transient hypothyroidism was diagnosed. We previously identified novel mutation p.A1206T in two unrelated patients with transient hypothyroidism [27]. This is compatible with findings of Moreno, who reported that biallelic mutations cause permanent hypothyroidism, whereas monoallelic mutations cause transient hypothyroidism [20]. However, studies have reported biallelic inactivating DUOX2 mutations in cases of mild hypothyroidism. Tonacchera identified a novel compound heterozygous mutation p.S911L/p.C1052Y in a child with CH and a eutopic thyroid gland [28]. Hoste [29] also reported a novel genetic defect (c.4552G>A, p.Gly1518Ser) together with deletion of exons 26–33 in a French-Canadian patient with transient CH and functional studies showed complete inactivation of DUOX2. Moreover, monoallelic permanent cases have indicated the heterogeneity of DUOX2 mutations and suggested the possible involvement of multiple gene defects or epigenetic mechanisms [30, 31]. De Marco et al. revealed a DUOX2 deletion mutation (p.S965fsX994) in three children with subclinical hypothyroidism and known functional impairment of H2O2 production, confirming that monoallelic mutations of DUOX2 protein can cause different phenotypes [30]. Muzza et al. found a p.S965fsX994 mutation in DUOX2 in a patient with permanent CH and PIOD [31]. These studies provide further evidence that the transient or persistent nature of the hypothyroid phenotype is not directly related to the number of inactivated DUOX2 alleles.
With the increasing number of reported DUOX2 mutations, genotype–phenotype correlations have become more complex than initially anticipated. The clinical spectrum of patients with DUOX2 mutation ranges from moderate to severe goitrous hypothyroidism. Yoshizawa-Ogasawara described two unrelated Japanese girls with a p.G488R mutation [32]. One patient was a compound heterozygote for p.L479SfsX3/p.G488R and the other was homozygous for p.G488R. This p.G488R substitution occurred in a highly conserved glycine residue of the mammalian DUOX2 protein. The two patients have different haplotypes, suggesting that the p.G488R alleles are the result of independent, recurrent mutations. Kasahara described a delayed-onset CH patient with two pathogenetic factors: a genetic defect and iodine excess [33]. Mutation screening for DUOX2 identified biallelic mutations (p.E327X/p.H678R). This provided a new example of environmental modification of the CH phenotypes by genetic defects, which can potentially distort screening results.
In conclusion, we have identified two missense DUOX2 mutations in Chinese patients with CH and goiter. In addition, we studied the functional significance of these mutants and revealed impaired H2O2 production and faster degradation in mutant DUOX2 proteins. Our findings confirm the genetic heterogeneity of DUOX2 mutations. However, further studies are required to elucidate the underlying mechanisms.
MATERIALS AND METHODS
Patients
Ten unrelated CH patients (four boys, six girls, sex ratio 1:1.5, age 2.8±1.2 years) were recruited through the neonatal screening program in Shandong Province, China, between 2008 to 2013. Blood samples were collected from the heel and TSH levels were measured by enzyme-linked immunosorbent assay according to the same test standard. Subjects with elevated TSH (≥20 μIU/ml) levels were recalled for further evaluation. Serum TSH (normal range 0.27–4.2 uIU/ml) and free thyroxin (FT4; normal range 12–22 pmol/L) were determined by electrochemiluminescence assay. CH is diagnosed by high serum TSH levels and low FT4 levels. Ten children were diagnosed with CH and goiter through a 99mTc thyroid scan or thyroid ultrasound examination. They underwent diagnostic reevaluation at 2–4 years of age, based on thyroid hormone testing after 1 month of L-T4 withdrawal. Patients showing persistently high TSH values were defined as having permanent CH. To confirm the diagnosis of transient or permanent CH, all subjects were routinely examined. Blood samples were collected from patients only after obtaining written informed consent from their guardians and the study was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University. All experiments were performed in accordance with the approved guidelines.
DNA analysis
Genomic DNA was extracted from peripheral blood leukocytes using standard methods. DUOX1, DUOXA1, DUOX2, TG and TPO mutation screening was negative in all 10 subjects. The human DUOX2 gene is encoded by 33 exons. Gene fragments covering the complete coding sequence, including intron-exon boundaries, of DUOX2 (GenBank NM_014080.4) were amplified using the appropriate primer pairs for exons 1–33 of the DUOX2, as reported previously [24]. Identical amplification conditions were used in a total volume of 25 μl containing 250 nM dNTPs, 100 ng of template DNA, 0.5 μM of each primer, and 1.25 U AmpliTaq Gold DNA polymerase, in 1x reaction buffer (10 mM Tris HCl, pH 8.3, 50 mM KCl, 2.5 mM MgCl2). The samples were denatured at 94°C for 5 min followed by 35 cycles consisting of 30s at 94°C, 1 min at 51–65°C and 30s at 72°C, and a final primer extension of 10 min at 72°C. Amplified PCR products were purified and sequenced using the appropriate PCR primers and the BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA), and run on an automated sequencer, ABI 3730XL (Applied Biosystems), for mutational analysis.
Construction of the expression vectors
cDNA was synthesized with reverse transcriptase M-MLV (Takara) by oligo(dT) priming of total RNA from a normal human thyroid gland. The DUOX2 and DUOXA2 open reading frames were amplified using native Pfu polymerase and cloned into pcDNA3.1 (Invitrogen). The primers for DUOX2 were 5′-GGGGTACCCCATGCTCCGTGCAAGACCAGAG-3′ and 5′-GCTCTAGAGCTCAGAAGTTCTCATAGTGGTGC-3′ and the primers for DUOXA2 were 5′-GGGGTACCCCATGACCCTGTGGAACGGCGTA-3′ and 5′-GCTCTAGAGCTCACAGGTTAGTGGTGATACA-3′. The two novel DUOX2 mutations p.R354W and p.A1206T were introduced into the DUOX2 expression vector by site-directed mutagenesis (Easy mutagenesis system, TransGen) using the following primers: p.R354W: F 5′-TGCCAGCTGTCATTTCTGGAAGGTCCT-3′ p.R354W:R 5′-AGAAATGACAGCTGGCATTTCTCATGTAG-3′ and p.A1206T: F5′-ATCATGTATGTCTTCACCTCCCACCA-3′ p.A120 6T:R5′-TGAAGACATACATGATGGCCAGGACC-3′. All constructs were confirmed by bidirectional DNA sequencing.
Cell culture and transfection
Before transfection, cells were divided into eight groups according to the expression vector to be transfected, including DUOX2 (containing DUOX2 wild-type (WT) expression vector only), exon 10 (containing R354W DUOX2 mutant expression vector only), exon 28 (containing A1206T DUOX2 mutant expression vector only), DUOX2 and DUOXA2 (containing DUOX2 WT expression vector and DUOXA2 WT expression vector), exon 10 and DUOXA2 (containing R354W DUOX2 mutant expression vector and DUOXA2 WT expression vector), exon 28 and DUOXA2 (containing A1206T DUOX2 mutant expression vector and DUOXA2 WT expression vector), pcDNA3.1(containing an empty expression vector), and non transfected WT cells (no expression vectors). Hela cells and 293T cells were maintained in high-glucose Dulbecco’s modified Eagle’s medium supplemented with 100 U/ml penicillin, 100 ug/ml streptomycin, and 10% fetal bovine serum under humidified conditions with 5% CO2 at 37°C. The diluted DNA (200 ng DUOX2 WT or DUOX2 mutants with or without 40 ng DUOXA2) and the diluted transfection reagent (with 0.5μl Lipofectamine TM2000 ) were combined into DNA-Lipofectamine TM2000 complexes at room temperature for 20 minutes. When Hela cells and 293T cells reached 70−80% confluence, the DNA-Lipofectamine™ 2000 complexes were added into each well containing cells and medium and incubated at 37°C in a CO2 incubator for 6 hours, after which culture solution was replaced by complete culture medium.
Measurement of H2O2 generation
Forty-eight hours after transfection, H2O2 production was determined in a reaction with cell-impermeable 10-acetyl-3,7-dihydroxyphenoxazine (Amplex Red reagent; Invitrogen) in the presence of excess peroxidase, producing fluorescent resorufin [34]. Briefly, cell monolayers were incubated in Dulbecco’s phosphate-buffered saline (PBS) supplemented with 50 mM Amplex Red reagent and 0.2 U/ml H2O2 for 1 hour at 37°C. Relative fluorescence (excitation/emission, 535/595 nm) of the medium was measured and corrected for Amplex Red oxidation in wells containing nontransfected cells. A calibration curve was used to convert changes in fluorescence intensity into absolute nanomoles and the H2O2 concentration was maintained within the linear range of the standard calibration curve throughout. Renilla luciferase activity from co-transfected pRL-Tk plasmid (Promega) was used as internal control [23].
Western blot analysis
Twenty-four hours after transfection, reaction termination was performed by 100 μg/mL cycloheximide (CHX; Sigma). Proteins were collected and extracted at different time intervals as follows: Cell pellets were washed once in PBS and resuspended in three volumes of CelLytic Lysis/Extraction Reagent (Sigma) with 1 mM PMSF. Cell suspension was incubated for 20 minutes on ice and cleared by centrifugation (12,000g, 15 minutes, 4°C). Total protein concentrations were determined by the Bradford method. Protein extracts were boiled in SDS/β-mercaptoethanol sample buffer. Samples (45 μg) were loaded into each lane of 8% polyacrylamide gels, separated by electrophoresis, and blotted onto polyvinylidene fluoride membranes (Amersham Pharmacia Biotech) by electrophoretic transfer. Western blotting was performed with a rabbit anti-DUOX2 antibody (Abgent) at 1:500 dilution, and bound antibody was detected using horseradish-peroxidase-conjugated goat anti-rabbit secondary IgGs. Detection by chemiluminescence reaction was performed using the ECL kit (Pierce, Appleton). Actin was detected as a loading control with a mouse anti-β-actin antibody (Santa Cruz). Three independent experiments were performed.
Statistical analysis
All data are expressed as mean ± standard deviation (SD) and were analyzed by one-way analysis of variance followed by Newman–Keuls multiple comparison test as appropriate (GraphPad Prism version 5 software). A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank all probands for their participation. This study was supported by the National Natural Science Foundation of China (81170812 and 81500689) and Science and Technology Project by Shandong Provincial Population and Family Planning Commission (No.5 in 2013). We also thank the various web sources including PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), and SIFT (http://sift.jcvi.org).
CONFLICTS OF INTEREST
The authors have no conflicts of interest relevant to this article to disclose.
Author contributions
S.L. and W.Z. drafted the initial manuscript. S.L.* and W.Z.* contribute equally to this work. S.Y. conceptualized and designed the study. L.Z. carried out the initial analyses. X.M. and H.X. reviewed and revised the manuscript. H.Z., K.L. and Q.L. designed the data collection instruments, and coordinated and supervised data collection at two of the four sites, critically reviewed the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
REFERENCES
1. Lorey FW, Cunningham GC. Birth prevalence of primary congenital hypothyroidism by sex and ethnicity. Human biology. 1992; 64:531-8.
2. Park SM, Chatterjee VK. Genetics of congenital hypothyroidism. Journal of medical genetics. 2005; 42:379-89. doi: 10.1136/jmg.2004.024158.
3. Chang WC, Liao CY, Chen WC, Fan YC, Chiu SJ, Kuo HC, Woon PY, Chao MC. R450H TSH receptor mutation in congenital hypothyroidism in Taiwanese children. Clinica chimica acta; international journal of clinical chemistry. 2012; 413:1004-7. doi: 10.1016/j.cca.2012.02.027.
4. Klett M. Epidemiology of congenital hypothyroidism. Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes Association. 1997; 105:19-23. doi: 10.1055/s-0029-1211926.
5. Targovnik HM, Citterio CE, Rivolta CM. Thyroglobulin gene mutations in congenital hypothyroidism. Hormone research in paediatrics. 2011; 75:311-21. doi: 10.1159/000324882.
6. Ris-Stalpers C, Bikker H. Genetics and phenomics of hypothyroidism and goiter due to TPO mutations. Mol Cell Endocrinol. 2010; 322:38-43. doi: 10.1016/j.mce.2010.02.008.
7. Spitzweg C, Morris JC. Genetics and phenomics of hypothyroidism and goiter due to NIS mutations. Mol Cell Endocrinol. 2010; 322:56-63. doi: 10.1016/j.mce.2010.02.007
8. Moreno JC, Visser TJ. New phenotypes in thyroid dyshormonogenesis: hypothyroidism due to DUOX2 mutations. Endocrine development. 2007; 10:99-117. doi: 10.1159/0000106822.
9. Zamproni I, Grasberger H, Cortinovis F, Vigone MC, Chiumello G, Mora S, Onigata K, Fugazzola L, Refetoff S, Persani L, Weber G. Biallelic inactivation of the dual oxidase maturation factor 2 (DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocr Metab. 2008; 93:605-10. doi: 10.1210/jc.2007-2020.
10. Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet. 1997; 17:411-22. doi: 10.1038/Ng1297-411.
11. Moreno JC, Visser TJ. Genetics and phenomics of hypothyroidism and goiter due to iodotyrosine deiodinase (DEHAL1) gene mutations. Mol Cell Endocrinol. 2010; 322:91-8. doi: 10.1016/j.mce.2010.03.010.
12. Corvilain B, van Sande J, Laurent E, Dumont JE. The H2O2-generating system modulates protein iodination and the activity of the pentose phosphate pathway in dog thyroid. Endocrinology. 1991; 128:779-85. doi: 10.1210/endo-128-2-779.
13. Ohye H, Sugawara M. Dual oxidase, hydrogen peroxide and thyroid diseases. Experimental biology and medicine. 2010; 235:424-33. doi: 10.1258/ebm.2009.009241.
14. Moreno JC, Pauws E, van Kampen AHC, Jedlickova M, de Vijlder JJM, Ris-Stalpers C. Cloning of tissue-specific genes using serial analysis of gene expression and a novel computational substraction approach. Genomics. 2001; 75:70-6. doi: 10.1006/geno.2001.6586.
15. Pachucki J, Wang D, Christophe D, Miot F. Structural and functional characterization of the two human ThOX/Duox genes and their 5 '-flanking regions. Mol Cell Endocrinol. 2004; 214:53-62. doi: 10.1016/j.mce.2003.11.026.
16. Dupuy C, Ohayon R, Valent A, Noel-Hudson MS, Deme D, Virion A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase - Cloning of the porcine and human cDNAs. J Biol Chem. 1999; 274:37265-9. doi: 10.1074/jbc.274.52.37265.
17. De Deken X, Wang DT, Many MC, Costagliola S, Libert F, Vassart G, Dumont JE, Miot F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem. 2000; 275:23227-33. doi: 10.1074/jbc.M000916200.
18. Caillou B, Dupuy C, Lacroix L, Nocera M, Talbot M, Ohayon R, Dème D, Bidart JM, Schlumberger M, Virion A. Expression of reduced nicotinamide adenine dinucleotide phosphate oxidase (ThoX, LNOX, Duox) genes and proteins in human thyroid tissues. J Clin Endocr Metab. 2001; 86:3351-8. doi: 10.1210/Jc.86.7.3351.
19. Rigutto S, Hoste C, Grasberger H, Milenkovic M, Communi D, Dumont JE, Corvilain B, Miot F, De Deken X. Activation of Dual Oxidases Duox1 and Duox2 DIFFERENTIAL REGULATION MEDIATED BY cAMP-DEPENDENT PROTEIN KINASE AND PROTEIN KINASE C-DEPENDENT PHOSPHORYLATION. J Biol Chem. 2009; 284:6725-34. doi: 10.1074/jbc.M806893200.
20. Moreno JC, Bikker H, Kempers MJE, van Trotsenburg P, Baas F, de Vijlder JJM, Vulsma T, Ris-Stalpers C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. New Engl J Med. 2002; 347:95-102. doi: 10.1056/Nejmoa012752.
21. Grasberger H. Defects of thyroidal hydrogen peroxide generation in congenital hypothyroidism. Mol Cell Endocrinol. 2010; 322:99-106. doi: 10.1016/j.mce.2010.01.029.
22. Jin HY, Heo SH, Kim YM, Kim GH, Choi JH, Lee BH, Yoo HW. High frequency of DUOX2 mutations in transient or permanent congenital hypothyroidism with eutopic thyroid glands. Hormone research in paediatrics. 2014; 82:252-60. doi: 10.1159/000362235.
23. Grasberger H, De Deken X, Miot F, Pohlenz J, Refetoff S. Missense mutations of dual oxidase 2 (DUOX2) implicated in congenital hypothyroidism have impaired trafficking in cells reconstituted with DUOX2 maturation factor. Mol Endocrinol. 2007; 21:1408-21. doi: 10.1210/me.2007-0018.
24. Varela V, Rivolta CM, Esperante SA, Gruneiro-Papendieck L, Chiesa A, Targovnik HM. Three mutations (p.Q36H, p.G418fsX482, and g.IVS19-2A > C) in the dual oxidase 2 gene responsible for congenital goiter and iodide organification defect. Clin Chem. 2006; 52:182-91. doi: 10.1373/clinchem.2005.058321.
25. Vigone MC, Fugazzola L, Zamproni I, Passoni A, Di Candia S, Chiumello G, Persani L, Weber G. Persistent mild hypothyroidism associated with novel sequence variants of the DUOX2 gene in two siblings. Human mutation. 2005; 26:395. doi: 10.1002/humu.9372.
26. Pfarr N, Korsch E, Kaspers S, Herbst A, Stach A, Zimmer C, Pohlenz J. Congenital hypothyroidism caused by new mutations in the thyroid oxidase 2 (THOX2) gene. Clinical endocrinology. 2006; 65:810-5. doi: 10.1111/j.1365-2265.2006.02672.x.
27. Wang F, Lu K, Yang Z, Zhang S, Lu W, Zhang L, Liu S, Yan S. Genotypes and phenotypes of congenital goitre and hypothyroidism caused by mutations in dual oxidase 2 genes. Clinical endocrinology. 2014; 81:452-7. doi: 10.1111/cen.12469.
28. Tonacchera M, De Marco G, Agretti P, Montanelli L, Di Cosmo C, Freitas Ferreira AC, Dimida A, Ferrarini E, Ramos HE, Ceccarelli C, Brozzi F, Pinchera A, Vitti P. Identification and functional studies of two new dual-oxidase 2 (DUOX2) mutations in a child with congenital hypothyroidism and a eutopic normal-size thyroid gland. The Journal of clinical endocrinology and metabolism. 2009; 94:4309-14. doi: 10.1210/jc.2009-0426.
29. Hoste C, Rigutto S, Van Vliet G, Miot F, De Deken X. Compound heterozygosity for a novel hemizygous missense mutation and a partial deletion affecting the catalytic core of the H2O2-generating enzyme DUOX2 associated with transient congenital hypothyroidism. Human mutation. 2010; 31:E1304-19. doi: 10.1002/humu.21227.
30. De Marco G, Agretti P, Montanelli L, Di Cosmo C, Bagattini B, De Servi M, Ferrarini E, Dimida A, Freitas Ferreira AC, Molinaro A, Ceccarelli C, Brozzi F, Pinchera A, et al. Identification and Functional Analysis of Novel Dual Oxidase 2 (DUOX2) Mutations in Children with Congenital or Subclinical Hypothyroidism. J Clin Endocr Metab. 2011; 96:E1335-E9. doi: 10.1210/jc.2010-2467.
31. Muzza M, Rabbiosi S, Vigone MC, Zamproni I, Cirello V, Maffini MA, Maruca K, Schoenmakers N, Beccaria L, Gallo F, Park SM, Beck-Peccoz P, Persani L, et al. The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. The Journal of clinical endocrinology and metabolism. 2014; 99:E544-53. doi: 10.1210/jc.2013-3618.
32. Yoshizawa-Ogasawara A, Ogikubo S, Satoh M, Narumi S, Hasegawa T. Congenital hypothyroidism caused by a novel mutation of the dual oxidase 2 (DUOX2) gene. Journal of pediatric endocrinology & metabolism : JPEM. 2013; 26:45-52. doi: 10.1515/jpem-2012-0082.
33. Kasahara T, Narumi S, Okasora K, Takaya R, Tamai H, Hasegawa T. Delayed onset congenital hypothyroidism in a patient with DUOX2 mutations and maternal iodine excess. American journal of medical genetics Part A. 2013; 161A:214-7. doi: 10.1002/ajmg.a.35693.
34. Mohanty JG, Jaffe JS, Schulman ES, Raible DG. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J Immunol Methods. 1997; 202:133-41. doi: 10.1016/S0022-1759(96)00244-X.