
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most frequent type of cancer in children and comprises neoplastic precursor cells committed to the B cell (BCP-ALL) or the T cell (T-ALL) lineages. BCP-ALL represents the majority of ALL, accounting for up to 85% of childhood ALL and 75% of adult ALL [1]. Genes involved in B cell development such as EBF1, IKZF1, PAX5 or PBX1 are frequently found mutated in BCP-ALL [2, 3]. Additionally, cytogenetic abnormalities are frequently detected and often define specific subtypes of the disease with unique prognostic features. While childhood BCP-ALL is curable in most cases with a survival rate approaching 80%, the rate of cure for adult patients presenting with BCP-ALL is only 40% [4, 5]. Despite progresses in the treatment of BCP-ALL, intensive chemotherapy regimens cause life threatening complication and around 20% of the treated patients relapse presenting then a very poor outcome [6]. There is a great need for the development of less toxic compounds and novel therapies for the treatment of relapsed and specific subgroups of BCP-ALL patients with poor prognosis.
The SOX family of transcription factor comprises 20 members divided into subgroups based on sequence similarities [7]. SOX factors are essential for the maintenance of various stem cell compartments during embryogenesis and adult life, such as for example SOX2 in neuronal stem progenitor cells [8] or SOX17 in fetal hematopoietic stem cells [9]. In addition, evidences for the involvement of SOX factors in cancers, either as tumour suppressors or as proto-oncogenes, have emerged recently. Deregulation of SOX gene expressions have been widely documented in cancer [10], and a few studies have identified a direct involvement of SOX factors in tumorigenesis [11–14]. SOX7 and its close homologues, SOX17 and SOX18, belong to the SOX F subgroup and play important roles in cardiovascular development [15–17]. The SOX F factors are also involved in the development of the hematopoietic system with SOX17 driving the expansion of fetal hematopoietic stem cells [9] while SOX7 and SOX18 are expressed at the earliest stages of blood development [18, 19]. The expression of SOX7 is frequently down-regulated in solid tumours such as prostate, colon and endometrial cancers, but the functional relevance of these findings remain unknown [20, 21]. A recent study has also revealed a tumor suppressor role for SOX7 in acute myeloid leukemia (AML) [22]. In the present study, we establish that SOX7 is frequently expressed in BCP-ALL and that the expression of this transcription factor is critical for BCP-ALL proliferation and maintenance.
RESULTS
SOX7 is frequently over-expressed in human BCP-ALL
To determine whether SOX7 expression was associated with hematological malignancies, we interrogated the Oncomine database for differential SOX7 expression in leukemias. This analysis revealed that SOX7 expression levels were frequently and significantly higher in childhood and adult BCP-ALL when compared to normal bone marrow and other types of leukemia, including T-ALL, acute myeloid leukemia (AML), chronic leukemias and myelodysplastic syndrome (MDS) (Figure 1A and S1A–S1B). In contrast, the expression of SOX17 and SOX18 did not show any specific or restricted pattern of expression in any hematological malignancies (Figure 1A). To validate this observation, the expression of SOX7 and SOX18 was evaluated in a small cohort of human ALL samples (Figure 1B–1C). These analyses confirmed that whilst SOX7 expression was high in multiple cases of BCP-ALL, SOX18 expression remained undetectable. Overall, SOX7 expression did not correlate with any specific type of chromosomal translocation or mutation known to be involved in BCP-ALL as confirmed by further analysis of the Oncomine datasets (Supplementary Figure S1C-S1D). Altogether, these results establish that the transcription factor SOX7 is specifically and frequently expressed in human BCP-ALL, without an obvious association with specific chromosomal abnormalities or cytogenetic subtypes.
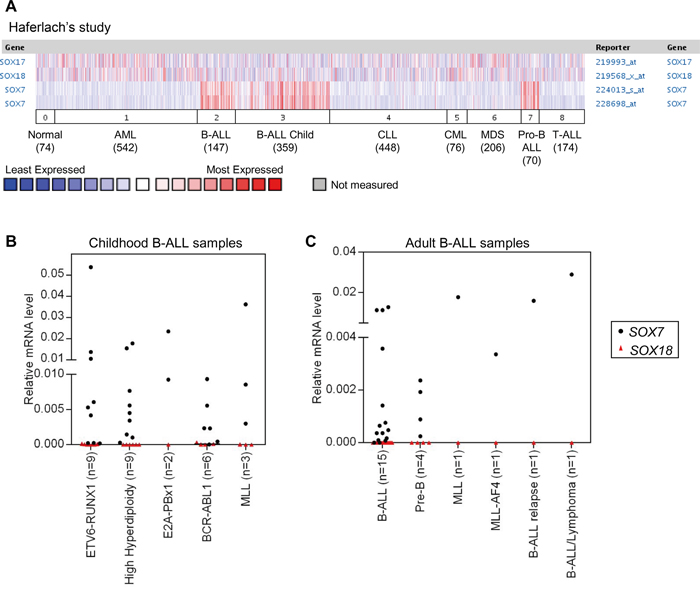
Figure 1: SOX7 is specifically expressed in human BCP-ALL samples. A. SOX7 expression dataset of human leukemia samples obtained from the Oncomine database. 0. Normal samples (74), 1. Acute Myeloid Leukemia (AML; 542), 2. B-Cell Acute Lymphoblastic Leukemia (BCP-ALL; 147), 3. B-Cell Childhood Acute Lymphoblastic Leukemia (BCP-ALL child; 359), 4. Chronic Lymphocytic Leukemia (CLL; 448), 5. Chronic Myelogenous Leukemia (CML; 76), 6. Myelodysplastic Syndrome (MDS; 206), 7. Pro-B Acute Lymphoblastic Leukemia (Pro BCP-ALL; 70), 8. T-Cell Acute Lymphoblastic Leukemia (T-ALL; 174). Two Affymetrix probes are shown for SOX7. B. Quantitative RT-PCR analysis of SOX7 and SOX18 transcript levels relative to β2-microglobulin in childhood human BCP-ALL samples (bone marrow). C. Quantitative RT-PCR analysis of SOX7 and SOX18 transcript levels relative to β2microglobulin in adult human BCP-ALL samples (blood).
SOX7 knock-down impairs the proliferation and clonogenicity of BCP-ALL cells
To investigate how SOX7 expression might contribute to the leukemic process, we first analyzed its expression in multiple human BCP-ALL cell lines with different cytogenetic characteristics (Figure 2A). The six lines evaluated expressed varying level of SOX7 transcript (Figure 2B) and protein (Figure 2C); MV4;11 an AML line used as a negative control did not expressed SOX7. To modulate SOX7 expression, we tested several shRNAs against SOX7 transcripts (Figure 2D), all of which induced greater than 95% decrease in SOX7 transcript levels in the endothelial HUVEC cells expressing high level of the SOX7 transcripts (Figure 2E). However, in BCP-ALL cells, these shRNAs induced much lower levels of knock-down. To evaluate the role of SOX7 in BCP-ALL, we therefore used two shRNAs conferring around 50% decrease in SOX7 transcript levels in RS4;11 and NALM6 expressing intermediate and high SOX7 level respectively (Figure 3A–3B). The two SOX7 shRNAs tested induced a significant decrease in proliferation in both leukemic cell lines when compared to the control shRNA (Figure 3C–3D). The co-transduction of two different SOX7 shRNAs in RS4;11 cells did not further decrease SOX7 transcript levels or proliferation when compared to cells transduced with a single shRNA (Supplementary Figure S2A-S2B). Cell cycle status via EdU incorporation revealed a reduction in the frequency of cells in the S-phase of the cell cycle for both leukemic cell lines transduced with either shRNAs (Figure 3E). Apoptosis assay showed that BCP-ALL cells were viable without significant increased apoptosis upon SOX7 knock-down (Figure 3F, positive control of staining Supplementary Figure S3). Next, to test whether SOX7 down-regulation impacted the clonogenic capacity of the BCP-ALL cells, we cultured the transduced cells in semi-solid colony assay, which represents a more stringent test of clonal proliferative capacity. The down-regulation of SOX7 by either shRNAs resulted in a significant decrease in colony formation in both leukemia lines (Figure 4A–4B), with much smaller colonies formed upon SOX7 knock-down (Figure 4C–4D). Altogether, these data demonstrate that the down-regulation of SOX7 in BCP-ALL human cell lines induced a significant decrease in proliferation, even though SOX7 transcript levels were only decreased by 50%.
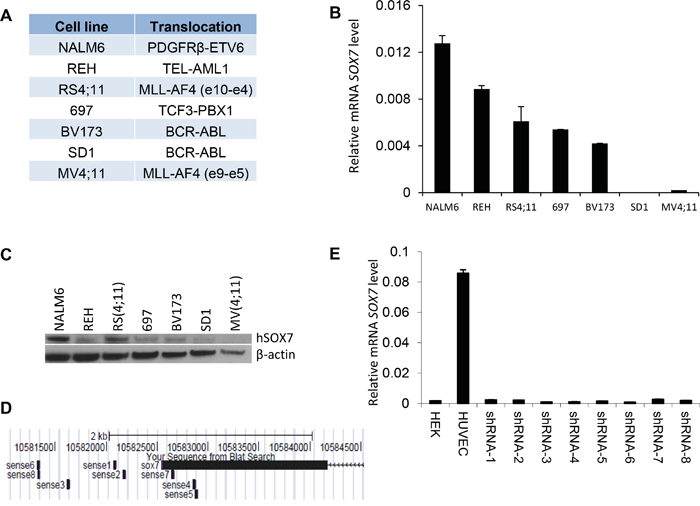
Figure 2: SOX7 expression in B-ALL cell lines. A. Human leukemia cell lines and their respective translocations. B. Quantitative RT-PCR analysis of SOX7 transcript level relative to β2microglobulin in leukemia cell lines. Error bars indicate mean ± standard deviation (n=3). C. Protein expression level for SOX7 and β-actin for the indicated cell lines measured by Western blot. Data are representative of two independent experiments. D. Binding regions of the different shRNAs to the human SOX7 locus; shRNAs are indicated as sense together with a number. Only shRNA 4, 5 and 7 are binding to SOX7 coding sequence, all the other shRNAs are binding to the UTR region. The alignment was performed using BLAST browser. E. Quantitative RT-PCR analysis of SOX7 transcript level relative to β2-microglobulin in HUVEC transduced with shRNA against SOX7 or not. HEK cells were used as negative control. Error bars indicate mean ± standard deviation (n=3).
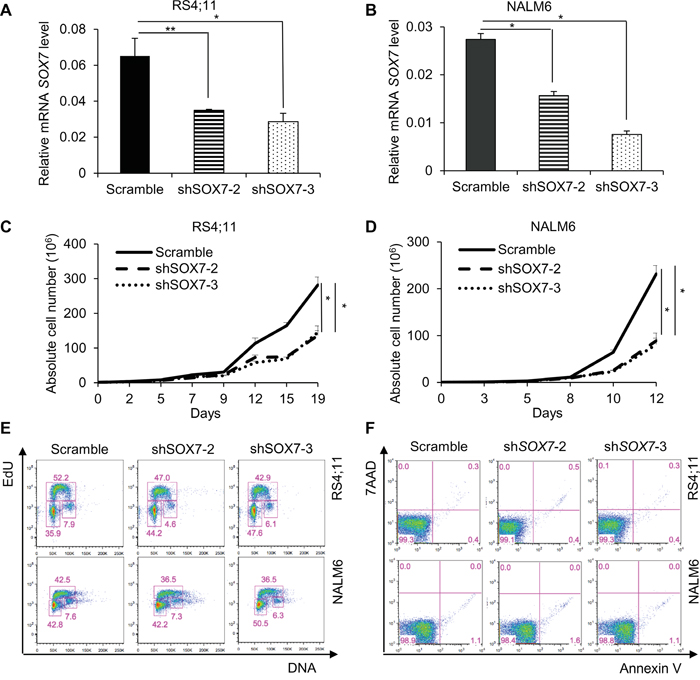
Figure 3: The down-regulation of SOX7 expression in BCP-ALL cells induces a significant decrease in proliferation. A-B. Quantitative RT-PCR analysis of SOX7 transcript levels relative to β2-microglobulin in RS4;11 and NALM6 human leukemia cells transduced with shRNA against SOX7 or control. Error bars indicate mean ± standard deviation (n=3; * P≤ 0.05; ** P<0.01). C-D. Representative line chart of cell count for RS4;11 and NALM6 cells transduced with shRNA against SOX7 or control cultured for 19 or 12 days respectively (* P<0.05). E. Cell cycle status of RS4;11 and NALM6 cells transduced with two different shRNAs against SOX7 (shSOX7-2, shSOX7-3) or control construct (Scramble). Staining was assessed after 2 hours of EdU incubation. Percentages of cells in the G0/G1, S and G2 phases are shown in the indicated gates. FACS plots are representative of two independent experiments. F. Representative FACS plots of 7AAD and Annexin-V expression profiles on RS4;11 and NALM6 cells transduced with two different shRNAs against SOX7 (shSOX7-2, shSOX7-3) or control construct (Scramble). Percentages of cells are shown in the indicated quadrants. FACS plots are representative of two independent experiments.
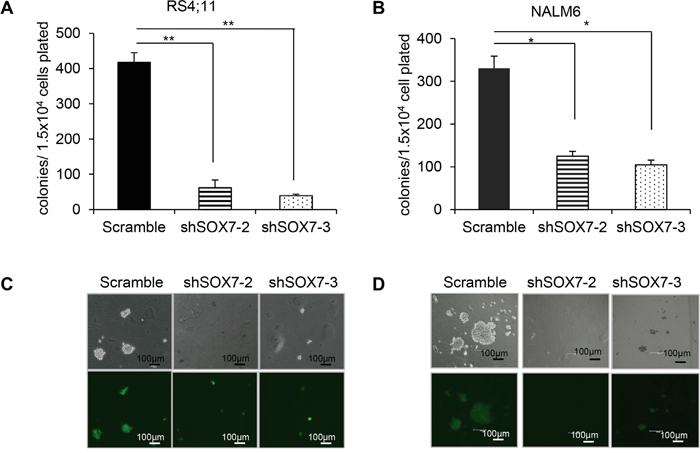
Figure 4: The down-regulation of SOX7 expression in BCP-ALL cells induces a significant decrease in clonogenicity. A-B. RS4;11 and NALM6 human leukemia cells transduced with the indicated shRNA were cultured in semi-solid clonogenic assays. Data represent colony number after 10 days in culture (n=3, * P<0.05; ** P<0.01). C-D. Representative pictures of colonies obtained from RS4;11 and NALM6 replating in colony assay at day 10. Scale bars represent 100μm (Leica scan and software). Data are representative of three independent experiments.
SOX7 knock-down delays leukemogenesis in vivo
To investigate the role of SOX7 in the progression of leukemia in vivo, recipient mice were engrafted either with RS4;11 leukemia cells transduced with a SOX7 or control shRNA or with NALM6 leukemia cells transduced with an inducible lentiviral vector encoding a SOX7 shRNA. An inducible system was tested to determine if acute deletion of SOX7 transcripts might be more efficient but similar results to the constitutive system were observed (Supplementary Figure S4A-S4B). Upon engraftment of the BCP-ALL control cells, recipient mice progressively developed symptoms indicative of leukemia, such as pale extremities, piloerection, hunched posture and continuous weight loss, ultimately leading to death. The knock-down of SOX7 expression in both cell lines induced a significant delay in the onset of these symptoms hence resulting in prolonged survival (Figure 5A–5B). A decrease in the leukemic burden was also observed post-mortem in the spleen of recipient mice engrafted with RS4;11 cells in which SOX7 expression was knocked-down (Figure 5C–5D). To further document leukemia development, in vivo imaging was performed for luciferase activity carried along the GFP reporter by the shRNA constructs (Figure 6A). Cells transduced with both shSOX7-2 and shSOX7-3 showed an overall similar level of GFP expression (hence luciferase) than the scramble control cells (Figure 6B), whereas cells transduced with a single SOX7 shRNA had lower GFP levels (hence luciferase) and therefore were not used for comparative imaging. Non-invasive in vivo luciferase imaging at the early stage of the disease clearly revealed a delay in the proliferation of BCP-ALL cells and in the spread of the leukemia through organ infiltration (Figure 6C). Taken together, these results demonstrate that the down-regulation of SOX7 delays the onset of BCP-ALL and decreases the leukemic burden, establishing the critical contribution of SOX7 expression in maintaining the high proliferative status of the leukemic cells.
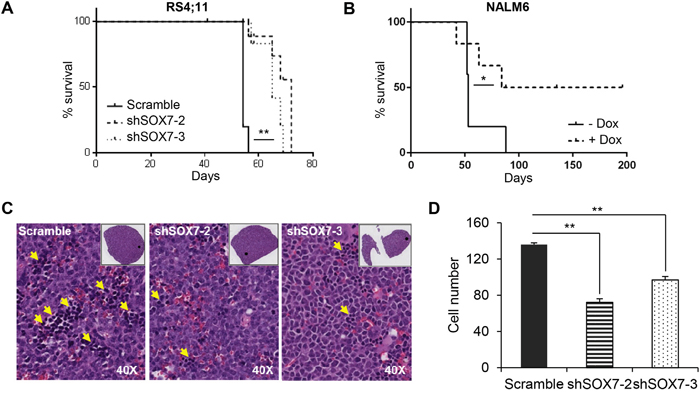
Figure 5: The down-regulation of SOX7 expression in BCP-ALL cells decreases leukemogenesis burden. A-B. Kaplan-Meier survival analysis. Mice were injected i.v. with RS4;11 human leukemia cells A. transduced with SOX7 or control shRNA or with NALM6 human leukemia cells B. transduced with an inducible SOX7 shRNA. Statistical differences between groups were calculated using the survival curve statistics Log-rank (Mantel-Cox) test (GraphPad Prism software). RS4;11 data are representative of three independent experiments (** P<0.05), NALM6 data represent two experiments (* P=0.08). (N=5 mice per group per experiment). C. May-Grünwald-Giemsa staining of spleen sections from mice engrafted with RS4;11 human leukemia cells transduced with two different SOX7 shRNAs (shSOX7-2 and shSOX7-3) or control construct (Scramble). Yellow arrowheads indicate leukemic cells. D. Bar chart representing the number of leukemic cells per section. Data are shown as the mean of cell number counted from four different areas in each section. Error bars indicate mean ± standard deviation (n=4, Leica scan and software, magnification: 40X). Data are representative of three independent experiments.
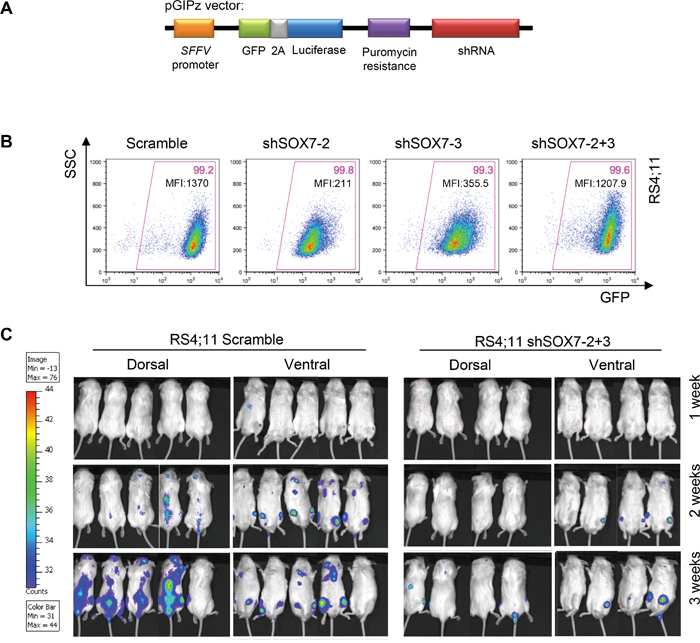
Figure 6: The down-regulation of SOX7 expression in BCP-ALL cells delays leukemogenesis initiation and spreading. A. Scheme of the pGIPz lentiviral vector used to transduce the leukemia cell lines. B. Representative FACS plots of SSC and GFP expression profiles of RS4;11 human leukemia cells transduced with SOX7 shRNAs (shSOX7-2, shSOX7-3, shSOX7-2+3) and control construct (Scramble). Percentages of cells and median fluorescent intensities are shown in the indicated quadrants. FACS plots are representative of two independent experiments. C. Mice were injected intra-peritoneally with Luciferin and anesthetized before to be imaged (IVIS Lumina, Caliper Life Sciences) for 30 seconds-1 minute per side (dorsal/ventral).
DISCUSSION
While specific recurrent cytogenetic abnormalities and mutations in key transcription factors of B cell lymphopoiesis have been identified in most BCP-ALL cases [1, 2], the molecular mechanisms underlying the emergence and maintenance of this type of leukemia still remain poorly understood. In the present study, we identify SOX7 as a novel and important player in BCP-ALL. We show that SOX7 is frequently and specifically expressed in BCP-ALL, independently of any specific chromosomal translocation, and that this transcription factor is implicated in the proliferative potential and clonogenicity of leukemic cells. Our study suggests that SOX7 expression in BCP-ALL is an important factor contributing to leukemia.
The analysis of SOX7 expression revealed the significant and specific expression of this transcription factor in BCP-ALL but not in other types of blood malignancy or in healthy bone marrow. Expression of SOX7 in BCP-ALL had no obvious association with specific chromosomal abnormalities or cytogenetic subtypes. Chromosomal translocations frequently found in BCP-ALL such as t(4;11) or t(12;21) producing the MLL-AF4 and ETV6-RUNX1 fusion proteins respectively, are not able on their own to fully recapitulate the human disease, suggesting that other critical events are required to lead to BCP-ALL [23–26], the deregulation of SOX7 expression could be one of these events. It remains to be determined however how SOX7 expression becomes deregulated, to date no chromosomal translocations or mutations have been identified within the SOX7 locus. An alternative possibility is that SOX7 is part of the transcriptional machinery regulating B cell development and as a result of mutational events leukemic cells become trapped in a SOX7-expressing stage of B lymphopoiesis in which SOX7 is a critical player of proliferation.
To investigate the role for SOX7 in leukemogenesis, we knocked-down the expression of SOX7 in human BCP-ALL cell lines using shRNAs. Eight different shRNA constructs were tested in HUVEC (endothelial cells) and all successfully and efficiently decreased SOX7 transcripts to less than 5% of the control levels. In contrast, in BCP-ALL cell lines, none of the shRNA constructs was able to decrease SOX7 transcript levels to less than 50% of the control levels. It is possible that this difference in knockdown efficiency is related to differences in the 3’UTR region of SOX7 mRNA between HUVEC and BCP-ALL cells as 3’UTR sequences are known to affect mRNA stability and post-transcriptional regulation [27]. Alternatively, it is possible that HUVECs do not rely on SOX7 expression for survival as much as BCP-ALL cell lines as HUVECs also express SOX17 and SOX18 that can control similar gene expression to SOX7 in endothelial cells [16]. Given that BCP-ALL cells do not express the two other SOXF factors, their survival might be compromised by a complete absence of SOX7 expression. However, even with this partial knock-down of SOX7, we observed a marked effect on proliferation and clonogenicity. It is likely that a more complete reduction in SOX7 level might result in more severe consequences in term of proliferation, survival or differentiation. Attempts to further decrease SOX7 transcript levels were investigated through transduction with two independent shRNAs or using an inducible shRNA system but none of these attempts was effective at further knocking-down SOX7 expression. These observations suggest that SOX7 expression is dispensable for the maintenance of HUVEC cells but that it is most likely critically required for BCP-ALL cell survival and proliferation.
Few SOX7 transcriptional targets have been identified to date and among them, we recently described VE-cadherin as one of SOX7 prime targets during the establishment of the cardiovascular system [28]. Interestingly, the expression of VE-cadherin has been reported in BCP-ALL cell lines and primary samples [29]. Furthermore, down-regulation of VE-Cadherin expression with siRNA or antagonist was shown to induce increase sensitivity to chemotherapy while VE-cadherin over-expression resulted in increased survival upon chemotherapy treatment [30]. Furthermore, the expression of endothelial adhesion molecules such as VE-cadherin has been linked to enhanced migration across the brain-blood endothelial barrier, promoting infiltration of the central nervous system by leukemic cells, an event contributing to relapse and poor outcome [31]. It is possible therefore that part of the leukemogenic potential of SOX7 might be achieved through its direct transcriptional activation of VE-cadherin. It will be important in future work to define the downstream program regulated by SOX7 in BCP-ALL and to identify the key pathways regulating the proliferative potential controlled by SOX7. They might represent potential novel druggable targets for the treatment of poor prognosis and relapsed BCP-ALL. Alternatively, the identification of small molecules that can interfere with SOX7 transcriptional activity either through SOX7-DNA interaction or through protein complex formation will represent interesting avenues of investigation with potential therapeutic benefits.
MATERIALS AND METHODS
Cell culture
RS4;11 human leukemia cells were grown in α-MEM (Lonza) supplemented with 10% FCS. NALM6 human leukemia cells were grown in RPMI (Lonza) supplemented with 10% FCS.
Western blotting
Protein samples were isolated using the RIPA lysis buffer and quantified followed by electrophoresis on 10% NuPAGE Novex Bis-Tris Mini Gels and blotting onto nitrocellulose membranes using Mini iBlot Gel Transfer Stacks Nitrocellulose (Invitrogen) Proteins were detected using human SOX7 antibody (R&D) and β-actin (Sigma).
Colony forming assay
RS4;11 and NALM6 human leukemia cells were plated in α-MEM or RPMI (both from Lonza) respectively containing 55% methylcellulose (10 g/L), 10% serum (Stem Cell Technology), 10% PFM (Gibco), 2 mM L-Glutamine (Gibco), 180 μg/ml transferrin, 0.5 mM ascorbic acid, 4.5x10-4 M MTG, 1% KL, 20 ng/ml IL-3, 10 ng/ml IL-7, 3 ng/ml IL-6 and 10 ng/ml SCF (all from PeproTech). Cultures were maintained at 37°C, 5% CO2. Colonies were counted after 10 days in culture according to their size and morphology.
Cell cycle analysis
106 cells were incubated with 10 μM EdU for 2 hours, harvested, fixed and permeabilized. EdU detection is based on a reaction between the azide, coupled to Alexa Fluor® 647 dye, and the alkyne in the EdU (Invitrogen). DNA content was measured using Violet406. Acquisition analysis was performed on a LSRII (BD cytometer).
Transplantation
RS4;11 or NALM6 human leukemia cells were transduced with lentiviruses for 30 minutes at 1200 rpm at MOI10-50. Transduced cells were sorted and transplanted i.v. into sub-lethally irradiated (125cGy) NSG mice. Leukemia growth was assessed by blood analysis, weight and general health monitoring. All animal work was performed under regulations governed by the Home Office Legislation under the Animal Scientific Procedures Act of 1986.
In vivo imaging
Mice were injected intra-peritoneally with Luciferin (75mg/kg; PerkinElmer), anesthetized (3% isofluorane) and imaged (IVIS, Caliper Life Sciences) for 1 minute per side.
Statistical analyses
Data were analyzed using a Student’s t Test. Significant differences are indicates with * (P<0.05) and ** (P<0.01). Survival analysis was performed using the Kaplan-Meier method and Log-rank statistical test.
ACKNOWLEDGMENTS
We thank Dr Tim Somervaille for scientific advices and input. We thank the staff at the Flow Cytometry, Advanced Imaging, Biological Research Unit and Molecular Biology Core facilities of CRUK Manchester Institute for technical support. Works in our laboratories are supported by Cancer Research UK (CRUK), Biotechnology and Biological Sciences Research Council (BBSRC) and Leukemia Lymphoma Research / Bloodwise. V.S. is a Wellcome-DBT India Alliance Margdarshi Fellow.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
Author contributions
S.C. designed and performed experiments, analyzed the data and wrote the manuscript. G.F. designed and performed experiments. A.M. and V.S. provided reagents and scientific input and advices. G.L. and V.K. designed and supervised the research project, analyzed the data and wrote the manuscript.
REFERENCES
1. Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, Ley TJ, Evans WE. The Pediatric Cancer Genome Project. Nat Genet. 2012; 44:619-622.
2. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007; 446:758-764.
3. Kastner P, Dupuis A, Gaub MP, Herbrecht R, Lutz P, Chan S. Function of Ikaros as a tumor suppressor in B cell acute lymphoblastic leukemia. American journal of blood research. 2013; 3:1-13.
4. Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004; 350:1535-1548.
5. Faderl S, O'Brien S, Pui CH, Stock W, Wetzler M, Hoelzer D, Kantarjian HM. Adult acute lymphoblastic leukemia: concepts and strategies. Cancer. 2010; 116:1165-1176.
6. Pui CH, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012; 120:1165-1174.
7. Schepers GE, Teasdale RD, Koopman P. Twenty pairs of sox: extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev Cell. 2002; 3:167-170.
8. Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003; 39:749-765.
9. Kim I, Saunders TL, Morrison SJ. Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell. 2007; 130:470-483.
10. Castillo SD, Sanchez-Cespedes M. The SOX family of genes in cancer development: biological relevance and opportunities for therapy. Expert Opin Ther Targets. 2012; 16:903-919.
11. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, Woo MS, Weir BA, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009; 41:1238-1242.
12. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, Brohee S, Salmon I, Dubois C, del Marmol V, Fuks F, Beck B, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014; 511:246-250.
13. Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014; 25:139-151.
14. Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, Zecchin D, Hobor S, Bajpe PK, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014; 508:118-122.
15. Francois M, Koopman P, Beltrame M. SoxF genes: Key players in the development of the cardio-vascular system. Int J Biochem Cell Biol. 2010; 42:445-448.
16. Hosking B, Francois M, Wilhelm D, Orsenigo F, Caprini A, Svingen T, Tutt D, Davidson T, Browne C, Dejana E, Koopman P. Sox7 and Sox17 are strain-specific modifiers of the lymphangiogenic defects caused by Sox18 dysfunction in mice. Development. 2009; 136:2385-2391.
17. Sakamoto Y, Hara K, Kanai-Azuma M, Matsui T, Miura Y, Tsunekawa N, Kurohmaru M, Saijoh Y, Koopman P, Kanai Y. Redundant roles of Sox17 and Sox18 in early cardiovascular development of mouse embryos. Biochem Biophys Res Commun. 2007; 360:539-544.
18. Gandillet A, Serrano AG, Pearson S, Lie ALM, Lacaud G, Kouskoff V. Sox7-sustained expression alters the balance between proliferation and differentiation of hematopoietic progenitors at the onset of blood specification. Blood. 2009; 114:4813-4822.
19. Serrano AG, Gandillet A, Pearson S, Lacaud G, Kouskoff V. Contrasting effects of Sox17- and Sox18-sustained expression at the onset of blood specification. Blood. 2010; 115:3895-3898.
20. Chan DW, Mak CS, Leung TH, Chan KK, Ngan HY. Down-regulation of Sox7 is associated with aberrant activation of Wnt/b-catenin signaling in endometrial cancer. Oncotarget. 2012; 3:1546-1556. doi: 10.18632/oncotarget.667.
21. Guo L, Zhong D, Lau S, Liu X, Dong XY, Sun X, Yang VW, Vertino PM, Moreno CS, Varma V, Dong JT, Zhou W. Sox7 Is an independent checkpoint for beta-catenin function in prostate and colon epithelial cells. Molecular cancer research. 2008; 6:1421-1430.
22. Man CH, Fung TK, Wan H, Cher CY, Fan A, Ng N, Ho C, Wan TS, Tanaka T, So EC, Kwong YL, Leung AY. Suppression of SOX7 by DNA methylation and its tumor suppressor function in acute myeloid leukemia. Blood. 2015; 125:3928-36. doi: 10.1182/blood-2014-06-580993.
23. Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, Kutok JL, Kung AL, Armstrong SA. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008; 14:355-368.
24. Zeisig BB, Cheung N, Yeung J, So CW. Reconstructing the disease model and epigenetic networks for MLL-AF4 leukemia. Cancer Cell. 2008; 14:345-347.
25. Fischer M, Schwieger M, Horn S, Niebuhr B, Ford A, Roscher S, Bergholz U, Greaves M, Lohler J, Stocking C. Defining the oncogenic function of the TEL/AML1 (ETV6/RUNX1) fusion protein in a mouse model. Oncogene. 2005; 24:7579-7591.
26. van der Weyden L, Giotopoulos G, Rust AG, Matheson LS, van Delft FW, Kong J, Corcoran AE, Greaves MF, Mullighan CG, Huntly BJ, Adams DJ. Modeling the evolution of ETV6-RUNX1-induced B-cell precursor acute lymphoblastic leukemia in mice. Blood. 2011; 118:1041-1051.
27. Matoulkova E, Michalova E, Vojtesek B, Hrstka R. The role of the 3' untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA biology. 2012; 9:563-576.
28. Costa G, Mazan A, Gandillet A, Pearson S, Lacaud G, Kouskoff V. SOX7 regulates the expression of VE-cadherin in the haemogenic endothelium at the onset of haematopoietic development. Development. 2012; 139:1587-1598.
29. Wang L, O'Leary H, Fortney J, Gibson LF. Ph+/VE-cadherin+ identifies a stem cell like population of acute lymphoblastic leukemia sustained by bone marrow niche cells. Blood. 2007; 110:3334-3344.
30. O'Leary H, Akers SM, Piktel D, Walton C, Fortney JE, Martin KH, Craig M, Coad J, Gibson LF. VE-cadherin Regulates Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia Sensitivity to Apoptosis. Cancer microenvironment. 2010; 3:67-81.
31. Akers SM, O'Leary HA, Minnear FL, Craig MD, Vos JA, Coad JE, Gibson LF. VE-cadherin and PECAM-1 enhance ALL migration across brain microvascular endothelial cell monolayers. Exp Hematol. 2010; 38:733-743.