
INTRODUCTION
Acute leukemia is the most frequent cancer in children. Although a remarkable improvement in survival of children with acute leukemia has been achieved, some patients still have a poor outcome. For these high-risk patients it is still necessary to find new biomarkers that may guide therapy or serve themselves as a target for new therapies. Among high-risk cases, infant leukemias (those diagnosed at age < 1 year) are a special subgroup with distinctive clinical and biological features, including frequent MLL (Mixed Lineage Leukemia, also known as KMT2A) rearrangements, high FLT3 (FMS-like tyrosine kinase 3) expression and high sensitivity to the nucleoside analogue cytarabine (Ara-C), but a dismal prognosis [1–6].
FLT3 is a tyrosine-kinase receptor with a key role in hematopoiesis. FLT3 mutations and overexpression have emerged as negative prognostic biomarkers in acute lymphoblastic leukemia (ALL) and acute myeloblastic leukemia (AML) [3, 7–10]. FLT3 is expressed in the majority of ALL and AML cases, but the highest levels have been observed in ALL with MLL gene rearrangements (ALL-MLL+) and hyperdiploid ALL (51–67 chromosomes, HeH) [6, 10]. FLT3 overexpression correlates with unfavorable prognosis in adult AML cases as well as in infant leukemias [8]. The overexpression of FLT3, even in the absence of activating mutations, causes phosphorylation of the receptor, resulting in a constitutively active form of FLT3 in a ligand-independent manner [3, 5, 8, 9, 11]. Internal tandem duplication of the FLT3 gene (FLT3-ITD) constitutively activates the receptor and confers a negative prognostic impact in both adult and pediatric AML cases, especially when a high mutant to wild-type allelic ratio is observed [3, 8, 12, 13].
Ara-C is a nucleoside analog that requires membrane transport proteins of the human nucleoside transporters (hNT) gene families SLC28 and SLC29 to be internalized into target cells. SLC28 encode human Concentrative Nucleoside Transporters (hCNT) whereas SLC29 encode human Equilibrative Nucleoside Transporter (hENT) proteins. Despite important advances in the knowledge of the tissue distribution and pharmacology of hNT proteins, there is still a limited understanding of the mechanisms that regulate their expression and activity [14]. Ara-C is known to be transported across the cell membrane mainly by Human Equilibrative Nucleoside Transporter 1 (hENT1, SLC29A1) [14, 15]. Once inside the cell, Ara-C undergoes metabolic activation, and the first and rate-limiting step in this process is its phosphorylation by deoxycitidine kinase (DCK), necessary to finally exert its cytotoxic action [16].
Uptake into cells is a key step in the bioavailability and pharmacological action of nucleoside analogues [16] and, accordingly, several studies have established a correlation between high hENT1 expression levels, drug sensitivity and outcome [14]. In this regard, elevated hENT1 expression has been reported to facilitate the high Ara-C sensitivity of infant ALL-MLL+, and a strong correlation between hENT1 levels and Ara-C sensitivity has been reported in those cases, as well as in adult AML patients [17–19]. On the contrary, low hENT1 expression levels have been related to Ara-C resistance in childhood AML. Overall, these data are in line with the evidence that in vitro Ara-C sensitivity in childhood and adult AML is dependent upon hENT1 [19].
In this setting, given that ALL-MLL+ cases present both high levels of FLT3 and high sensitivity to Ara-C, we hypothesized that FLT3 is a suitable candidate to modulate hNT expression and activity, thereby contributing to cell chemosensitivity. To address this issue, we analyzed the relationship between FLT3 expression levels and mutations and the expression and activity of different hNT and Ara-C metabolizing enzymes in different cell lines and in a series of ALL and AML pediatric patients.
RESULTS
FLT3 is highly expressed in pediatric ALL-MLL+ patients
FLT3 mRNA expression was quantified by RQ-PCR in 50 patients and 3 cell lines (MV4-11, SEM, K562) and normalized against mRNA from commercial bone marrow CD34+ cells. The expression of FLT3 was heterogeneous, with a median (arbitrary units) of 4.35 (0.09–4470). Among the different cytogenetic subgroups, the highest FLT3 levels were observed in ALL-MLL+ patients; none of them had FLT3-ITD (data not shown). We found no significant differences between FLT3 levels and gender, CNS status or white blood cell count.
Positive correlation between hENT1 and FLT3 mRNA expression in pediatric leukemia samples
The mRNA amounts of the main nucleoside transporters (NT) and intracellular metabolizing enzymes (ME), hENT1, hENT2 (SLC29A2), hCNT1 (SLC28A1), hCNT2 (SLC28A2), hCNT3 (SLC28A3), DCK and cN-II (NT5C2), were measured in 50 pediatric leukemia cases by RQ-PCR. When correlating FLT3 mRNA levels with all these genes, noticeably, despite the high interpatient variability, a positive correlation was found between FLT3 and hENT1 mRNA levels (Figure 1). Based upon these observations we could hypothesize that samples with high FLT3 mRNA levels would show high Ara-C uptake and probably, a better response to therapy. On the other hand, no significant differences were found between hENT1, hENT2, hCNT1, hCNT2, and hCNT3, DCK and cN-II mRNA expression levels and age, gender, CNS status and WBC count. Nevertheless, a positive correlation between FLT3 and DCK was also observed (Supplementary Figure S1).
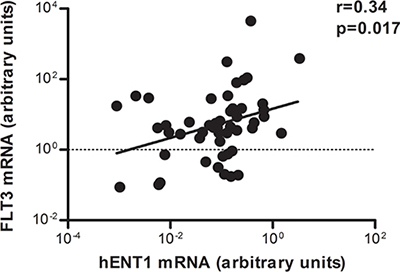
Figure 1: Correlation between hENT1 and FLT3 mRNA expression in pediatric leukemia samples. Relative hENT1 mRNA levels of cells from 50 pediatric patients with acute leukemia were plotted against the levels of FLT3 mRNA in the same samples. Correlation coefficient is shown in the figure.
Although our study was not designed to analyze NT or ME expression patterns in acute leukemia molecular subgroups, we also found significantly higher hENT1 levels in AML-MLL+ and lower DCK and cN-II levels in AML cases and T-ALL samples.
Characterization of the mRNA expression and associated biological function of nucleoside transporters in leukemic cell lines
As nucleoside transporter expression in pediatric leukemic cells is not well known we analyzed the mRNA expression levels of the most relevant NT that could potentially be implicated in Ara-C uptake, hENT1, hENT2, hCNT1 and hCNT3, in MV4-11, SEM and K562 leukemic cell lines. The former two cell lines originated from childhood leukemia. We observed that hENT1, hENT2 and hCNT1 expression is retained in the three cell lines, whereas barely detectable hCNT3 mRNA was found in SEM and K562 cell lines (Supplementary Table S1).
Knowing that all cell lines expressed almost all NT mRNAs, we then aimed at determining the transportability profiles for each cell line. For this purpose, we performed radiolabeled cytidine uptake measurements. We chose cytidine because it is a pyrimidine that it is known to be a good substrate for hENTs, hCNT1 and hCNT3, all the putative Ara-C transporters. All cell lines showed hENT1-related function (Figure 2A) which was found to be the predominant nucleoside transporter activity in the three cell lines. We also detected, albeit to a lesser extent, hENT2 and hCNT1-related activities in all of them. MV4- 11 also retained some hCNT3-related function, being this activity undetectable in K562 and SEM cells.
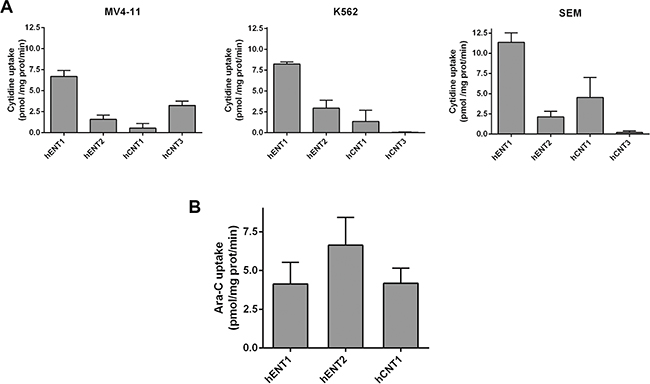
Figure 2: Characterization of nucleoside transporter activity implicated in Ara-C uptake. (A) [3H]-cytidine uptake (1 μM, 1 min) by MV4-11, K562 and SEM cells and (B) [3H]-Ara-C transport (1 μM, 1min) in MV4-11 cell line were assayed. Cross-inhibitions were performed by adding to the transport medium cold guanosine (100 μM) and inhibition of hENT1 was achieved adding NBTI (1 μM) to the transport medium. Data are expressed as means ± SEM of triplicate measurements from three independent cultures.
Nucleoside transporters implicated in Ara-C uptake
Direct transport of Ara-C by each of the previously identified NTs was determined. Given that the only cell line that retained all the NT-related activities was MV4- 11, we decided to focus on this cell line to study Ara-C transport. We observed that Ara-C was translocated inside cells by hENT1, hENT2 and hCNT1 (Figure 2B).
FLT3 effect on hENT1 expression and activity
As we previously detected a positive correlation between FLT3 and hENT1 expression in the cohort of primary samples from pediatric patients we decided to evaluate the role FLT3 plays in modulating hENT1 expression. For this purpose we initially analyzed the effects of the FLT3 inhibitor, PKC412, on MV4-11 and SEM cell lines. All NTs and ME mRNA levels were determined in MV4-11 and SEM cells either in the presence or in the absence of PKC412 used at a concentration based upon previous literature [20]. Acute treatment with PKC412 resulted in a significant down-regulation of hENT1-related mRNA levels at the first studied time-point (8 h) without any further change at 24 h after treatment (Figure 3A). No changes in hENT2-related mRNA levels were found under the same conditions (Figure 3B). The down-regulation of the hENT1-related mRNA levels resulted in decreased hENT1 protein amounts (Figure 3C). Overall these data suggest that down-regulation of hENT1 is somehow specific and occurs immediately after PKC412 treatment no matter whether activated FLT3 is the result of its overexpression (SEM cells) or the consequence of mutations resulting in the constitutive activation of this kinase (MV4-11). Under the experimental conditions described above, it was also demonstrated that PKC412 inhibited FLT3 phosphorylation, without affecting total FLT3 expression (Figure 3D) proving that the inhibitor was acting as expected. To further assess the involvement of FLT3 in hENT1 regulation, another FLT3 inhibitor likely to be more specific than PKC412, AC220, was also used. AC220 could also inhibit FLT3 phosphorylation, thereby inducing a significant down-regulation of hENT1 protein expression (Supplementary Figure S2).
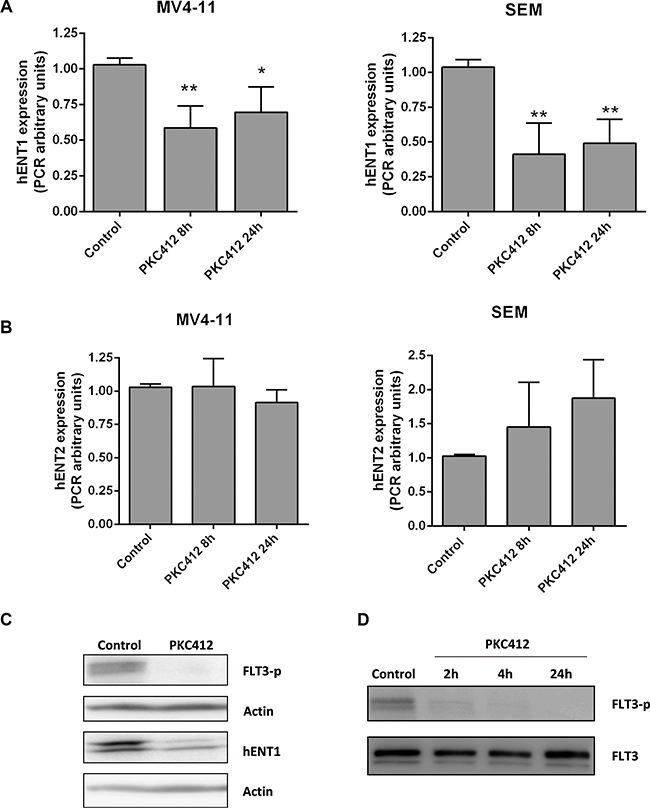
Figure 3: Involvement of FLT3 in hENT1 expression. mRNA expression of (A) hENT1 and (B) hENT2 were assayed by quantitative RQ-PCR in MV4-11 and SEM cells treated for 8 or 24 h with FLT3 inhibitor PKC412 (0.045 μM). Results are based on duplicate measurements from three independent experiments. Western blot for hENT1 (C), FLT3 and phospho-FLT3 (Tyr591) (D) were performed in cell extracts from MV4-11 cells incubated in the presence of PKC412 (0.045 μM) for different periods of time. A representative Western blot of three independent experiments is shown in each panel. Statistical significance denotes significant difference relative to control cells (*p < 0.05; **p < 0.01).
The functional impact of hENT1 down-regulation following FLT3 inhibition was demonstrated by monitoring hENT1-mediated cytidine uptake in MV4- 11 cells. hENT1 function was significantly decreased reaching its maximum inhibition at 16 h incubation with PKC412 inhibitor (Supplementary Figure S3).
Effect of FLT3 inhibition on Ara-C uptake and cytotoxicity
Considering that FLT3 inhibition induced the down-regulation of hENT1 mRNA expression and related activity, without significantly affecting other NTs, we then aimed at determining the effect of FLT3 inhibition on Ara-C transport. For this purpose, we cultured MV4- 11 cells with PKC412 for 16 h and evaluated direct Ara-C uptake mediated by the previously demonstrated entities that were responsible for the transport of the drug, hENT1, hENT2 and hCNT1. Under conditions of maximal FLT3 inhibition, hENT1-mediated Ara-C uptake was dramatically decreased (Figure 4), without significantly affecting hENT2 and hCNT1 related activities. However, the residual hENT1 activity was still significant, representing nearly a 25% of that of untreated cells. In any case, no compensatory up-regulation associated with the other Ara-C transporters was observed.
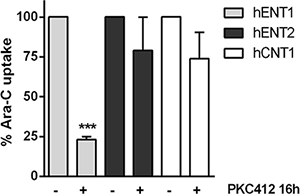
Figure 4: Effect of FLT3 inhibition in the uptake of Ara-C. Direct uptake of [3H]Ara-C (1 μM, 1 min) was measured in MV4-11 cells in the presence of PKC412 for 16 h either in medium containing NaCl or choline chloride. Sodium-dependent transport was calculated as uptake in NaCl medium minus uptake in choline chloride medium. Data are normalized to uptake measured in the absence of PKC412 and expressed as percentage ± SEM from 3 independent experiments, each conducted in quadruplicate. Statistical significance denotes significant difference relative to control cells (***p < 0.001).
To address the question of whether Ara-C cytotoxicity could be affected by hENT1 down-regulation under FLT3 inhibition conditions, cytotoxicity assays were performed. Ara-C was used at IC50 concentrations in SEM and MV4-11 cells (not shown). Under conditions of hENT1 activity inhibition (using 1 μM NBTI) Ara-C induced cytotoxicity was significantly blocked, whereas addition of phloridzin (an hCNT inhibitor) did not significantly alter Ara-C action (Figure 5A). Thus, in basal conditions (no PKC412 present), Ara-C induced cytotoxicity was mostly related to hENT1 function. When MV4-11 cells were treated with the FLT3 inhibitor PKC412 for 16 h, followed by a 6 h exposure to Ara-C, impact on cell survival was not significantly greater than when treating the cells with PKC412 alone (Figure 5B); however, if the exposure to the drugs was the opposite (6 h Ara-C treatment, followed by a 16 h exposure to PKC412), this combined treatment resulted in a significantly greater cell death than when treating the cells with PKC412 alone (Figure 5C). Thus, a lower cytotoxicity was observed if PKC412 was given previous to Ara-C exposure, probably due to a reduction in hENT1 activity. In contrast, once Ara-C had been administered, the subsequent addition of PKC412 resulted in greater cytotoxicity. In the former experimental design, the calculation of the coefficient of drug interaction (CDI) revealed the occurrence of drug antagonism (CDI: 1.20), whereas in the latter design, the effect of both drugs turned out to be additive (CDI: 1.05). Interestingly, when these experiments were performed using the FLT3 inhibitor AC220, similar results were obtained (Supplementary Figure S4), thereby reinforcing the key role of FLT3 in hENT1 modulation and cytarabine-associated cytotoxicity.
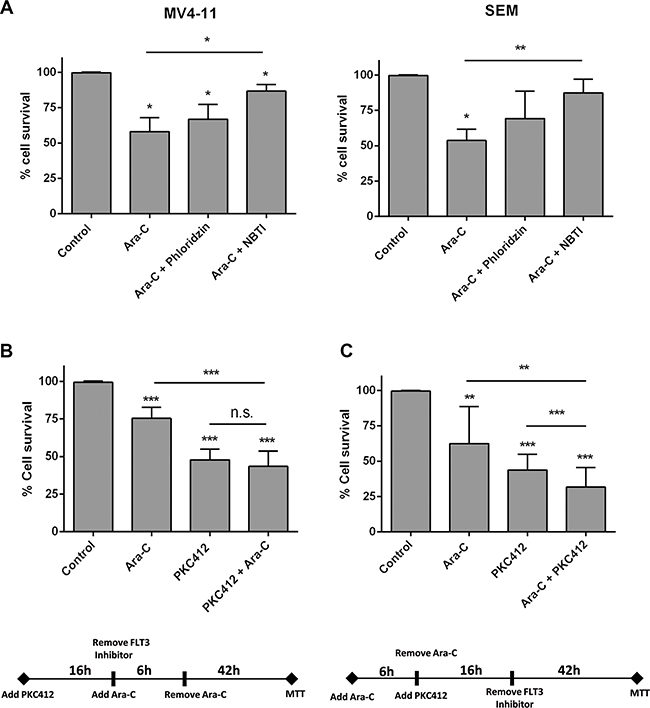
Figure 5: Effect of FLT3 inhibition in the cytotoxicity induced by Ara-C. Cell viability was determined by MTT assays when (A) MV4-11 and SEM cells were cultured with Ara-C (3 μM and 1 μM for MV4-11 and SEM cells respectively) either in the presence or not of NBTI (1 μM) or phloridzin (250 μM) for 48 h; (B) MV4-11 cells were treated with the FLT3 inhibitor PCK412 (16 h), followed by a 6 h exposure to Ara-C (10 μM), and (C) MV4-11 cells were cultured with Ara-C (10 μM) for 6 h, followed by a 16 h exposure to PKC412. Data are expressed as percentage of survival ± SEM of triplicate measurements from six-nine independent experiments. Statistical significance denotes significant difference relative to control cells (*p < 0.1; **p < 0.01; ***p < 0.001) or to Ara-C or PKC412 treated cells as indicated.
DISCUSSION
FLT3 mutations have emerged in the last years as key prognostic biomarkers in AML. FLT3-ITD mutation is a recognized adverse prognostic factor for outcome in adult and pediatric AML patients [2, 3, 5, 7, 8, 21–23]. Accordingly, patients harboring FLT3- ITD mutations are considered as high-risk patients, and benefit from hematopoietic stem cell transplant (HSCT) in first complete remission (CR1) [2, 21, 24]. FLT3 overexpression is associated with FLT3 phosphorylation, thereby resulting in constitutive activation of the receptor, similarly to FLT3 mutants [3, 5, 8, 12]. Some authors have correlated FLT3 overexpression, in absence of FLT3 mutation, with poor outcome in both ALL and AML [4, 5, 8]. Beyond these clinical observations, little is known about the exact biological mechanism that confers high risk in such patients. Armstrong and co-authors [25] revealed that FLT3 gene overexpression is the most distinctive feature of MLL rearranged acute leukemia. Moreover, a significantly higher FLT3 expression level was observed in infant ALL-MLL+ patients. Interestingly, infant patients are highly sensitive to Ara-C, but show in vitro resistance to prednisone and asparaginase [1]. All these data have been taken into account to design the chemotherapeutic schemes in the International Cooperative Treatment Protocol for Infants with ALL, INTERFANT (99 & 06) [26].
Ara-C is mainly, but not exclusively, transported by hENT1, being this plasma membrane transporter a major contributor to Ara-C bioavailability and, likely, action. In fact, high hENT1-related mRNA levels have been reported in ALL-MLL+ patients, and this evidence may explain, at least partially, their Ara-C sensitivity [17]. The same group demonstrated that MLL rearrangements were no directly involved in Ara-C sensitivity in infant ALL and childhood AML-MLL+ [30], pointing out that other mechanisms are involved in Ara-C sensitivity in these patients.
In our study, the analysis of a cohort of 50 pediatric leukemia patients revealed a positive correlation between the expression (mRNA levels) of FLT3 and hENT1. These results would be consistent with previous observations showing that subgroups of patients with high sensitivity to cytarabine frequently overexpress FLT3 [3, 10, 27] . Our results showing high hENT1 and FLT3 expression and a positive correlation between both parameters suggest that FLT3 might play a relevant role on Ara-C sensitivity, likely to involve hENT1 up-regulation.
We could not find any significant association between FLT3 expression levels and other clinical or biological features, although this analysis was far out of the scope of our study. Moreover, a bias related to the selection of determined molecular subtypes in our cohort of patients was also introduced during the design of our study.
mRNA levels of other putatives Ara-C transporters (hENT2, hCNT1, hCNT3) and those of the intracellular metabolizing enzymes DCK and cN-II were also determined in the same cohort of patients, although no correlations among NT and ME and clinical and biological parameters were observed. Previous studies have suggested a correlation between specific NT expression profiles and outcome in solid tumors and hematological malignancies [15, 19, 28, 29], being this issue recently reviewed by ourselves [14]. However, the limited number of patients within the present cohort did not allow us to establish prognostic correlations.
We also found a positive correlation between FLT3 and DCK expression (Supplementary Figure S1), the kinase involved in the first phosphorylation step of nucleoside analogs once they have entered the cells via NTs. Low levels of DCK tend to correlate with Ara-C resistance in childhood ALL and AML and adult AML [18, 30, 31]. Our observations suggest that DCK would not be a limiting step in Ara-CTP formation in patients with high FLT3 expression and thus, would support intracellular activation (i.e. phosphorylation) of Ara-C. These results would be in agreement with other studies [19] showing an in vitro correlation between hENT1 and DCK expression and nucleoside analogs sensitivity in pediatric and adult AML [18]. Recently, a collaborative EORTC-GIMEMA trial has demonstrated a better response rate and survival with high dose Ara-C in very high risk setting patients, including AML FLT3-ITD cases [22], that presumably would show high hENT1 expression according to our results.
Nucleoside analogues are the mainstay treatment in AML and are broadly used in relapsed-refractory ALL salvage schedules with diverse success rate [32–36]. As we here demonstrate a significant correlation between FLT3 and Ara-C transporter hENT1, we propose that FLT3 expression could be used as a nucleoside analogue sensitivity surrogate.
Not surprisingly, several FLT3 inhibitors have been developed in the last few years, and some of them are currently used in advanced-phases clinical trials with variable success rate (https://clinicaltrials.gov/). FLT3 inhibitors are basically expected to inhibit cellular proliferation through the down-regulation of this kinase, thus resulting in a withdrawal of antiapoptotic signals [37–39]. Some FLT3 inhibitors revealed higher efficacy when combined with chemotherapy [38, 40–42]. However, the best schedule combination with conventional chemotherapy has not been identified yet and research using different approaches to overcome inhibitors resistance is still needed. Given that a significant correlation was found in our study between FLT3 expression and hENT1 expression, we also wanted to explore how FLT3 inhibitors could modulate Ara-C uptake and related cytotoxicity. We verified that Ara-C is transported by hENT1 as it was previously reported, and we also found that Ara-C can also be transported into the cells by hENT2 and hCNT1. We next demonstrated that the FLT3 inhibitor PKC412 induced a significant reduction of hENT1 mRNA expression in both MV4-11 and SEM cell lines, which results in functional down-regulation of hENT1-dependent drug uptake. Similar effects on hENT1 down-regulation were found with AC220, a more selective FLT3 inhibitor. This effect appears to be specific, as PKC412 does not modify other transporter functions (i.e. hENT2 and hCNT1). Taken together, our results suggest that Ara-C transporter hENT1 expression and activity are regulated by FLT3. Interestingly, Jin and colleagues [20], using heterologous transduction of FLT3- ITD in HF6 cells, had suggested that FLT3-ITD down-regulates hENT1 expression in a heterologous system using transduced FLT3-ITD in HF6 cells. Oppositely, our results achieved in endogenously expressing FLT3 cell lines (wild type FLT3 and FLT3-ITD) suggest that FLT3 is indeed up-regulating hENT1 expression and related activity. Those results are fully consistent with the clinical data, showing a positive correlation between FLT3 and hENT1 mRNA expression levels in the cohort of 50 patients here analyzed.
Regarding the effect on cell survival of the combination of a FLT3 inhibitor and a chemotherapeutic drug, two studies [43, 44] have previously demonstrated in vitro that combined therapies involving FLT3 inhibitors and chemotherapeutical agents might yield variable efficacy depending upon the sequence of drug treatment. Thus, antagonistic cytotoxic effects were induced when they pretreated FLT3-ITD cell lines and primary patient samples with a FLT3 inhibitor followed by chemotherapy. Similar results were obtained in wild type FLT3 with high FLT3 mRNA expression levels. This effect appears to be the result of the FLT3 inhibitor inducing cell cycle arrest at the G1-S phase, thereby perturbing the action of genotoxic drugs, such as Ara-C. The possibility of FLT3 modulating hENT1 as a result of cell cycle perturbation should not be ruled out, although it looks unlikely to us. hENT1 has been shown to provide extracellular nucleosides for DNA synthesis in murine primary bone marrow macrophages [45], but hCNT1 is the transporter known to be cell cycle-dependent, showing up-regulation at the S phase [46]. Moreover, whereas FLT3 inhibition induces down-regulation of hENT1 it does not change at all either hCNT1 mRNA expression levels or hCNT1-related activity. In summary, we think FLT3-related hENT1 modulation might not be the indirect result of cell cycle arrest induced by FLT3 inhibition. However, our data do further support the relevance of the sequence of drug treatment. In fact, under conditions of hENT1 down-regulation, the cytotoxic effect triggered by either PKC412 or AC220 was not further potentiated by Ara-C treatment, whereas this was not the case when cells were first treated with Ara-C, and basal hENT1 activity had not been yet down-regulated by FLT3 inhibition. Under these conditions the cytotoxic efficacy of the combined therapy was significantly much greater than when treating the cells with the FLT3 inhibitor alone. This finding highlights also the need for a better understanding of the basal regulatory properties of drug transporters, which may be targets of kinase inhibitors used in combined therapies.
In summary, we have demonstrated that Ara-C transport into cells is mediated by hENT1, hENT2 and hCNT1. More importantly, we also demonstrated that FLT3 significantly regulates hENT1 expression and activity and thereby Ara-C sensitivity. We have also proved that the sequence of the administration of FLT3 inhibitors and Ara-C is important, as the cytotoxic efficacy of the latter is greater when FLT3 inhibitors follow Ara-C administration. Overall, our data might contribute to better understand how FLT3 may influence drug sensitivity and to develop new therapeutic approaches in order to improve the overall outcome of patients with high risk subtypes of leukemia.
MATERIALS AND METHODS
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the authors’ institutional review board.
Patient samples and cell lines
Among 265 pediatric patients aged 0–18 years diagnosed with acute leukemia from 2003 to 2013 in Hospital Sant Joan de Déu, we selected 50 cases (B-cell precursor ALL: 44; T-ALL: 2; AML: 4) with available biological samples for the study. We selected cases with the main genetic abnormalities reported in ALL but, as we wanted to determine the possible influence of FLT3 over nucleoside transport activity, we conducted a bias towards cases with expected high FLT3 expression, that is, patients with ALL and hyperdyploidy (51-67 chromosomes, HeH) and MLL rearranged cases (ALL-MLL+). The main clinical and biological characteristics of patients are described in Table 1. Patients were all uniformly treated according to the Spanish Society of Pediatric Hematology and Oncology (SEHOP) consecutive protocols SHOP-LAL-99 & 05 (ALL cases) and SHOP-LAM-00 & 07 (AML cases). We used normal bone marrow CD34+ cells as calibrator for FLT3 and hNT expression. According to the Local Ethics Committee of our institution, all samples were stored in the legally competent Biobank of our Hospital and were used after informed consent was obtained either from the patients or their legal tutors.
Table 1: Clinical and biological characteristics of the 50 patients diagnosed with acute leukemia included in our study
N = 50 |
|
|---|---|
Age, years (range) |
4.3 (0–16) |
Gender, n (%) |
|
Male |
26 (52) |
Female |
24 (48) |
CNS, n (%) |
|
CNS1 |
46 (92) |
CNS2ta |
3 (6) |
CNS3 |
1 (2) |
WBC count, ×109/L, median (range) |
17.6 (1.1–331.2) |
Hemoglobin, g/L, median (range) |
7.7 (2.9–11.7) |
Platelets, ×109/L, median (range) |
52 (2–520) |
Blasts, median (range) |
|
Bone marrow |
93 (58–100) |
Peripheral blood |
55 (0–99) |
Immunophenotype, n (%) |
|
Precursor B-ALL |
44 (88) |
T-ALLb |
2 (4) |
AML |
4 (8) |
Genetics, n (%) |
|
ALL: |
|
HeH |
19 (38) |
Otherc |
16 (32) |
ETV6-RUNX1 |
4 (8) |
MLL+ |
3 (6) |
TCF3-PBX1 |
2 (4) |
BCR-ABL1 |
2 (4) |
AML: |
|
MLL+ |
3 (6) |
Otherd |
1 (2) |
a: CNS2t: traumatic lumbar puncture.
b: Two cases of Early T-cell Precursor T-ALL, one of them harboring a FLT3-ITD mutation, were included.
c: Other B-cell precursor ALL cases (n = 14) included patients with normal karyotype (n = 5), cases with < 20 assessable metaphases (n = 6), cases with abnormalities at chromosome 9p (n = 3).
The two T-cell ALL cases included had a normal karyotype.
d: Other AML cases: we included a patient with 47, XX, t(6;9)(p23;q34), + 13[6]/46, XX, t(6;9)(p23;q34)[6] harboring a FLT3-ITD mutation.
Note: CNS: central nervous system. WBC: white blood cell. PB: peripheral blood. ALL: acute lymphoblastic leukemia. AML: acute myeloblastic leukemia. HeH: high hyperdiploid ALL (51–67 chromosomes).
We analyzed 3 different acute leukemia cell lines, SEM (childhood B-cell precursor ALL with translocation t(4;11) and MLL rearrangement with demonstrated high FLT3 expression and no FLT3 mutation; DSMZ ACC 546); MV4-11 (childhood AML with translocation t(4;11) and MLL rearrangement, harboring a FLT3-ITD mutation; DSMZ ACC 102) and K562 (lymphoid blast crisis of chronic myeloid leukemia with known low FLT3 expression and no FLT3 mutations; DSMZ ACC 10). K562 and MV4-11 cells were routinely cultured in RPMI-1640 medium (Lonza, Walkersville, MD) supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS) (Life Technologies, Carlsbad, CA), 50 μg/ml penicillin-streptomycin (Invitrogen, Carlsbad, CA), and 2 mM L-glutamine (Invitrogen, Carlsbad, CA). SEM cells were cultured in Iscove’s Modified Dubecco’s Medium (IMDM) (Gibco, Breda, Netherlands) similarly supplemented with 10% heat-inactivated FBS, 50 μg/ml penicillin-streptomycin and 2 mM L-glutamine.
DNA and RNA extraction and reverse transcription
Bone marrow or peripheral blood samples from diagnosis were used for the analysis. Separation of mononuclear cells was performed using density gradient with Ficoll-Hypaque (Sigma, St Louis MO, USA) and DNA was extracted with Qiaquick DNA extraction kit (Qiagen, Hilden, Germany). Total RNA was extracted with TriPure (Roche Diagnostics, Indianapolis, IN) and cDNA synthesis was performed from 10 ng of total RNA, using the QuantiTect Whole Transcriptome Kit (Qiagen, Hilden, Germany).
FLT3 mutational status
FLT3-ITD was studied by amplification of the juxtamembrane domain spanning exons 14 and 15, using fluorescently-labeled primers and subsequent analysis on a 3130XL Genetic Analyzer (Applied Biosystems, Life Technologies, Foster City, CA, USA), as previously described [47]. To detect point mutations in codon D835 or deletions within codon I836, we amplified the exon 20 of FLT3 and digested the product with Eco-RV enzyme, as reported [48]. All positive cases were directly sequenced to confirm the presence of mutations, using the BigDye Terminator Cycle Sequencing Kit v3.0 (Applied Biosystems, Life Technologies).
Quantification of FLT3 mRNA and protein expression
The quantification of the FLT3 gene-related mRNA was performed by real-time quantitative PCR (RQ-PCR) using the TaqMan® Gene Expression Assay Hs00174690_m1 (Applied Biosystems, Life Technologies) (see Supplementary Table S2), in a Light-Cycler 480 II (Roche Diagnostics, Indianapolis, IN), according to the manufacturer′s instructions. Relative quantification was calculated with the 2−ΔΔCt method, using β-glucuronidase (GUS) (NM_000181.2, ref. 4310888E) as endogenous gene and normal bone marrow CD34+ cells as calibrator.
FLT3 protein levels before and after treatment with the FLT3 inhibitors PKC412 (Enzo Life Science, Plymouth Meeting, PA) and AC220 (Selleck Chemicals, Houston, TX) were quantified by Western blot. For this purpose, cells were treated with either PKC412 (0.045 μM) or AC220 (0.5 nM) and lysed in a buffer containing 20 nM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% triton X-100 supplemented with 1 mM sodium orthovanadate, protease inhibitor (Complete mini; Roche, Basel, Switzerland) and phosphatase inhibitor cocktails (PhosSTOP; Roche). Fifty μg of proteins were separated by SDS-PAGE on standard 10% gel and transferred to Immobilon-P membranes (Millipore, Berford, MA, USA). Membranes were incubated with FLT3 (8F2) and phospo-FLT3 (Tyr591) antibodies (Cell Signalling, MA, USA). After washing with TBS-Tween, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. Immunoreactive bands were detected by chemiluminiscence (ECL, Amersham Pharmacia Biotech, NJ, USA).
Quantification of NT and metabolizing enzymes
Real-time quantitative PCR (RQ-PCR) amplification of hCNTs and hENTs were performed with primers and probes from Applied Biosystems, summarized in Supplementary Table S2, using the TaqMan Universal Master Mix, 700 nmol/L probe and 150 nmol/L of each primer in the ABI Prism 7700 Sequence Detection System (Applied Biosystems). The relative mRNA level of each gene was calculated with the 2−ΔΔCt method and normalized to that of β-glucuronidase (GUS) expression level and normal bone marrow CD34+ cells as calibrator. Absolute RQ-PCR results were obtained from interpolation of ΔCT of each sample in line regression standards for number of cDNA copies of each gene. hENT1 protein levels were semi-quantified by Western Blot, using a commercial anti-hENT1 polyclonal antibody (STJ96396) from Saint John’s Laboratory Ltd. (London, UK).
hNT activity assays
Nucleoside uptake was measured in MV4-11, SEM and K562 cells using a method adapted from a technique previously described by our laboratory [49]. Cells were washed twice and resuspended in either a 137 mM NaCl or 137 mM choline chloride buffer. Uptake assays were started by mixing cell suspensions with a 10% of the final volume of the same buffer, supplemented with a radionucleoside - [3H]Ara-C or [3H]cytidine – at a final concentration of 1 μM (specific activity 1 μCi/nmol) at a specific activity of 4000 dpm/pmol. Incubation was stopped after 1min (linear initial velocity conditions) by washing the cells three times in 1 mL of a cold buffer composed of 137 mM NaCl and 10 mM Tris (hydroxymethyl) aminomethane-HEPES (pH 7.4). Cells were then dissolved in 1 ml Triton-X-100 and aliquots were sampled for protein determination, according to Bradford (Bio-Rad, Hercules, CA), and for radioactivity measurements.
In the presence of Na+, hENTs and hCNTs are functional, although only hCNTs require this ion for substrate translocation. Thus, sodium dependent nucleoside transport activity (hCNT-related) was determined by subtracting uptake rates measured in the choline chloride medium (almost exclusively related to hENT1 and hENT2 activities) from measurements obtained in the sodium containing buffer (in which both ENT and CNT are active).
Cross-inhibition experiments were performed as described above, but adding saturating concentrations (100 μM) of a secondary non-radiolabeled nucleoside to the incubation media, which will compete for the transporter and block the transport of radiolabeled substrate. By adding extra guanosine to [3H]cytidine uptake media, hCNT3 but not hCNT1 activity will be blocked. hCNT1-mediated uptake was determined by substracting the activity measured in [3H]cytidine + guanosine conditions from [3H]cytidine alone and hCNT3 related activity from the substraction of [3H]cytidine + guanosine transport activity from the one determined when using [3H]cytidine alone. The equilibrative (Na+-independent) transport component inhibited by NBTI (1 μM) accounts for the hENT1-related nucleoside transport activity, whereas the NBTI-resistant transport includes residual hENT2-related uptake plus diffusion and binding components, which in general are negligible.
Cell treatment and apoptosis detection by MTT assay
For routine cytotoxicity assays, 2 × 104 cells were cultured either in the presence or the absence of appropriate inhibitors. To unveil the role selected nucleoside transporters can play in cytarabine-induced cytotoxicity cells were incubated for 15 min either in the presence or in the absence of NBTI (1 μM for hENT1 inhibition) or phloridzin 250 μM (for hCNT1inhibition). Afterwards Ara-C was added (3 μM and 1 μM for MV4-11 and SEM cells respectively) and cytotoxicity determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT; Sigma-Aldrich) after 48 h. Optical density (OD) was measured at 550 nm. For the analysis of combined drug effects cells were cultured with either 0.045 μM PKC412 or 0.5 nM AC220 and 10 μM Ara-C, following the experimental design described in the Results section, aimed at elucidating the probable role of the order of drug administration of chemotherapeutic efficacy. The Coefficient of Drug Interaction (CDI) was calculated as explained in [50].
Statistical analysis
For the correlation of clinical and biological variables, we used the χ2 and the Fisher′s exact tests for categorical variables and the Student′s t-test or the Mann–Whitney U-test for non-parametric tests. The Spearman non-parametric test was used to determine correlations among mRNA expression values of the analyzed genes. In addition, a Kruskal–Wallis test was used to compare the gene expression levels of FLT3 and NT among the different ALL subgroups. As we selected certain subtypes of leukemia according to their expected FLT3 expression and also the total number of primary cases is low, we did not perform survival analysis in our study. All p values were considered significant when < 0.05. All the statistical analyses were performed using the SPSS 22.0 statistical software package (SPSS Inc., Chicago, IL, USA).
ACKNOWLEDGMENTS
We are indebted to patients and families for their help and to Obra Social from Hospital Sant Joan de Déu for their support. We are indebted to the “Biobanc de l′Hospital Infantil Sant Joan de Déu per a la Investigació” integrated in the Spanish Biobank Network of ISCIII for the sample and data procurement.
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
GRANT SUPPORT
This study has been funded by projects PI12/2417 (AC, RM, SR, NVG, MC), integrated in Plan Nacional de I+D+I and cofunded by Instituto de Salud Carlos III (ISCIII) – Subdirección General de Evaluación y Fomento de la Investigación Sanitaria – and Fondo Europeo de Desarrollo Regional (FEDER) (AC, RM, SR, NVG, MC, LCC, MPA, PFC) and SAF2011-23660 and SAF2014-52067-R (MPA) by grants from Asociación Española Contra el Cáncer AECC-Foundation (AC, SR, MC), Fundación Sandra Ibarra de Solidaridad Frente al Cáncer (RM, SR, MC), Fundación Cris contra el Cáncer (MC), “Força Miquel” ,“Mua” & “Candela, polsera solidària” projects (AC, RM, SR, NVG, MC). CIBER is an initiative of ISCIII (Madrid, Spain). LCC is being funded by the Becas Chile Program (CONICYT, Gobierno de Chile).
REFERENCES
1. Pieters R, den Boer ML, Durian M, Janka G, Schmiegelow K, Kaspers GJ, van Wering ER, Veerman AJ. Relation between age, immunophenotype and in vitro drug resistance in 395 children with acute lymphoblastic leukemia—implications for treatment of infants. Leukemia. 1998; 121344–8.
2. Hasle H. A critical review of which children with acute myeloid leukaemia need stem cell procedures. Br J Haematol. 2014; 16623–33.
3. Stam RW, den Boer ML, Schneider P, Meier M, Beverloo HB, Pieters R.D-HPLC analysis of the entire FLT3 gene in MLL rearranged and hyperdiploid acute lymphoblastic leukemia.Haematologica. 2007; 921565–8.
4. Stam RW, Schneider P, de Lorenzo P, Valsecchi MG, den Boer ML, Pieters R. Blood. Blood. United States. 2007; 2774–5.
5. Kang H, Wilson CS, Harvey RC, Chen I-M, Murphy MH, Atlas SR, Bedrick EJ, Devidas M, Carroll AJ, Robinson BW, Stam RW, Valsecchi MG, Pieters R, et al. Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children’s Oncology Group study.Blood. 2012; 1191872–81.
6. Yeoh E-J, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A, Cheng C, Campana D, Wilkins D, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling.Cancer Cell. 2002; 1133–43.
7. Staffas A, Kanduri M, Hovland R, Rosenquist R, Ommen HB, Abrahamsson J, Forestier E, Jahnukainen K, Jónsson G, Zeller B, Palle J, Lönnerholm G, Hasle H, et al. Nordic Society of Pediatric Hematology and Oncology (NOPHO). Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood. 2011; 1185905–13.
8. Ozeki K, Kiyoi H, Hirose Y, Iwai M, Ninomiya M, Kodera Y, Miyawaki S, Kuriyama K, Shimazaki C, Akiyama H, Nishimura M, Motoji T, Shinagawa K, et al. Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood. 2004; 1031901–8.
9. Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, van der Voort E, Valsecchi MG, de Lorenzo P, Sallan SE, Armstrong SA, Pieters R. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood. 2005; 1062484–90.
10. Andersson A, Paulsson K, Lilljebjörn H, Lassen C, Strömbeck B, Heldrup J, Behrendtz M, Johansson B, Fioretos T. FLT3 mutations in a 10 year consecutive series of 177 childhood acute leukemias and their impact on global gene expression patterns.Genes Chromosomes Cancer. 2008; 4764–70.
11. Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, Pieters R, Kersey JH, Sallan SE, Fletcher JA, Golub TR, Griffin JD, Korsmeyer SJ. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification.Cancer Cell. 2003; 3173–83.
12. Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood. 2005; 105812–20.
13. Pratcorona M, Brunet S, Nomdedéu J, Ribera JM, Tormo M, Duarte R, Escoda L, Guàrdia R, Queipo de Llano MP, Salamero O, Bargay J, Pedro C, Martí JM, et al. Grupo Cooperativo Para el Estudio y Tratamiento de las Leucemias Agudas Mieloblásticas.Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood. 2013; 1212734–8.
14. Pastor-Anglada M, Pérez-Torras S. Nucleoside transporter proteins as biomarkers of drug responsiveness and drug targets.Front Pharmacol. 2015; 613.
15. White JC, Rathmell JP, Capizzi RL. Membrane transport influences the rate of accumulation of cytosine arabinoside in human leukemia cells. J Clin Invest. 1987; 79380–7.
16. Errasti-Murugarren E, Pastor-Anglada M. Drug transporter pharmacogenetics in nucleoside-based therapies.Pharmacogenomics. 2010; 11809–41.
17. Stam RW, den Boer ML, Meijerink JPP, Ebus MEG, Peters GJ, Noordhuis P, Janka-Schaub GE, Armstrong SA, Korsmeyer SJ, Pieters R. Differential mRNA expression of Ara-C-metabolizing enzymes explains Ara-C sensitivity in MLL gene-rearranged infant acute lymphoblastic leukemia.Blood. 2003; 1011270–6.
18. Abraham A, Varatharajan S, Karathedath S, Philip C, Lakshmi KM, Jayavelu AK, Mohanan E, Janet NB, Srivastava VM, Shaji RV, Zhang W, Abraham A, Viswabandya A, et al. RNA expression of genes involved in cytarabine metabolism and transport predicts cytarabine response in acute myeloid leukemia. Pharmacogenomics. 2015; 16877–90.
19. Hubeek I, Stam RW, Peters GJ, Broekhuizen R, Meijerink JPP, van Wering ER, Gibson BES, Creutzig U, Zwaan CM, Cloos J, Kuik DJ, Pieters R, Kaspers GJL. The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br J Cancer. 2005; 931388–94.
20. Jin G, Matsushita H, Asai S, Tsukamoto H, Ono R, Nosaka T, Yahata T, Takahashi S, Miyachi H. FLT3-ITD induces ara-C resistance in myeloid leukemic cells through the repression of the ENT1 expression. Biochem Biophys Res Commun. 2009; 3901001–6.
21. Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, Kaspers GJL, Heerema NA, Gerbing R, Lange BJ, Radich JP. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006; 1083654–61.
22. Willemze R, Suciu S, Meloni G, Labar B, Marie JP, Halkes CJM, Muus P, Mistrik M, Amadori S, Specchia G, Fabbiano F, Nobile F, Sborgia M, et al. High-dose cytarabine in induction treatment improves the outcome of adult patients younger than age 46 years with acute myeloid leukemia: results of the EORTC-GIMEMA AML-12 trial. J Clin Oncol. 2014; 32219–28.
23. Braoudaki M, Karpusas M, Katsibardi K, Papathanassiou CH, Karamolegou K, Tzortzatou-Stathopoulou F. Frequency of FLT3 mutations in childhood acute lymphoblastic leukemia.Med Oncol. 2009; 26460–2.
24. Brunet S, Labopin M, Esteve J, Cornelissen J, Socié G, Iori AP, Verdonck LF, Volin L, Gratwohl A, Sierra J, Mohty M, Rocha V. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. J Clin Oncol. 2012; 30735–41.
25. Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002; 3041–7.
26. Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, Hovi L, LeBlanc T, Szczepanski T, Ferster A, Janka G, Rubnitz J, Silverman L, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007; 370240–50.
27. Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, Sallan SE, Korsmeyer SJ. FLT3 mutations in childhood acute lymphoblastic leukemia.Blood. 2004; 1033544–6.
28. Gloeckner-Hofmann K, Guillén-Gómez E, Schmidtgen C, Porstmann R, Ziegler R, Stoss O, Casado FJ, Rüschoff J, Pastor-Anglada M. Expression of the high-affinity fluoropyrimidine-preferring nucleoside transporter hCNT1 correlates with decreased disease-free survival in breast cancer. Oncology. 2006; 70238–44.
29. Fernández-Calotti PX, Colomer D, Pastor-Anglada M.Translocation of nucleoside analogs across the plasma membrane in hematologic malignancies. Nucleosides Nucleotides Nucleic Acids. 2011; 301324–40.
30. Galmarini CM, Thomas X, Graham K, El Jafaari A, Cros E, Jordheim L, Mackey JR, Dumontet C.Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival in acute myeloid leukaemia patients treated with cytarabine. Br J Haematol. 2003; 12253–60.
31. Kakihara T, Fukuda T, Tanaka A, Emura I, Kishi K, Asami K, Uchiyama M. Expression of deoxycytidine kinase (dCK) gene in leukemic cells in childhood: decreased expression of dCK gene in relapsed leukemia. Leuk Lymphoma. 1998; 31405–9.
32. Locatelli F, Moretta F, Rutella S. Management of relapsed acute lymphoblastic leukemia in childhood with conventional and innovative approaches. Curr Opin Oncol. 2013; 25707–15.
33. Byrne JL, Dasgupta E, Pallis M, Turzanski J, Forman K, Mitchell D, Haynes AP, Russell NH. Early allogeneic transplantation for refractory or relapsed acute leukaemia following remission induction with FLAG. Leukemia. 1999; 13786–91.
34. McCarthy AJ, Pitcher LA, Hann IM, Oakhill A. FLAG (fludarabine, high-dose cytarabine, and G-CSF) for refractory and high-risk relapsed acute leukemia in children.Med Pediatr Oncol. 1999; 32411–5.
35. Hijiya N, Barry E, Arceci RJ. Clofarabine in pediatric acute leukemia: current findings and issues. Pediatr Blood Cancer. 2012; 59417–22.
36. Tavil B, Aytac S, Balci YI, Unal S, Kuskonmaz B, Yetgin S, Gurgey A, Tuncer M, Gumruk F, Uckan D, Cetin M. Fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin (FLAG-IDA) for the treatment of children with poor-prognosis acute leukemia: the Hacettepe experience. Pediatr Hematol Oncol. 2010; 27517–28.
37. Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, Jones-Bolin S, Ruggeri B, Dionne C, Small D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002; 993885–91.
38. Stubbs MC, Armstrong SA. FLT3 as a therapeutic target in childhood acute leukemia. Curr Drug Targets. 2007; 8703–14.
39. Konig H, Levis M. Targeting FLT3 to treat leukemia. Expert Opin Ther Targets. 2015; 1937–54.
40. Grunwald MR, Levis MJ. FLT3 inhibitors for acute myeloid leukemia: a review of their efficacy and mechanisms of resistance. Int J Hematol. 2013; 97683–94.
41. Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005; 10554–60.
42. Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, Murphy KM, Dauses T, Allebach J, Small D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004; 1033669–76.
43. Levis M, Pham R, Smith BD, Small D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004; 1041145–50.
44. Brown P, Levis M, McIntyre E, Griesemer M, Small D. Combinations of the FLT3 inhibitor CEP-701 and chemotherapy synergistically kill infant and childhood MLL-rearranged ALL cells in a sequence-dependent manner. Leukemia. 2006; 201368–76.
45. Soler C, García-Manteiga J, Valdés R, Xaus J, Comalada M, Casado FJ, Pastor-Anglada M, Celada A, Felipe A. Macrophages require different nucleoside transport systems for proliferation and activation. FASEB J. 2001; 151979–88.
46. Valdés R, Casado FJ, Pastor-Anglada M. Cell-cycle-dependent regulation of CNT1, a concentrative nucleoside transporter involved in the uptake of cell-cycle-dependent nucleoside-derived anticancer drugs. Biochem Biophys Res Commun. 2002; 296575–9.
47. Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M, Neubauer A, Ehninger G, Illmer T. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002; 994326–35.
48. Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002; 1002393–8.
49. Fernández-Calotti P, Pastor-Anglada M. All-trans-retinoic acid promotes trafficking of human concentrative nucleoside transporter-3 (hCNT3) to the plasma membrane by a TGF-beta1-mediated mechanism. J Biol Chem. 2010; 28513589–98.
50. Urtasun N, Vidal-Pla A, Pérez-Torras S, Mazo A. Human pancreatic cancer stem cells are sensitive to dual inhibition of IGF-IR and ErbB receptors. BMC Cancer. 2015; 15:223.