
INTRODUCTION
The kidney is essential for excretion of waste and maintenance of proper osmotic and oncotic pressure; these functions require a properly functioning filtration unit comprised of the glomerulus and surrounding Bowman’s capsule. Blood is filtered within the filtration unit; protein, cells, and specific concentrations of chemicals are retained, whereas waste and extra fluid is passed to the renal tubules to become urine. One common feature of glomerular dysfunction is loss of protein from the blood (hypoproteinemia) accompanied by increased urinary protein (proteinuria). Glomerular dysfunction can cause chronic kidney disease and eventually end-stage renal disease (ESRD), requiring dialysis or kidney transplantation. Various causes include genetic mutations, autoimmunity, infections, environmental exposures or any combination thereof (reviewed in [1-4]). Pathological changes damaging the glomerular filtration apparatus are responsible for 90% of ESRD, resulting in ~$20 billion in yearly health costs in the USA [5].
The glomerular filtration unit has three layers: the visceral epithelial cells (podocytes), the glomerular basement membrane, and parietal endothelial cells; proper communication and function of all three components is required for glomerular function. Podocytes, the most essential cell-type for glomerular filtration, possess interdigitating foot processes that form filtration slits called slit diaphragms (reviewed in [6]). Loss of the slit diaphragm through effacement of the podocyte foot processes is sufficient to cause proteinuria and kidney damage, emphasizing the importance of podocytes in proper kidney function [7]. Various podocytopathies play a dominant role in glomerular diseases [2].
Some cancer chemotherapeutic agents, including alkylating agents, induce nephrotoxicity. For example, oxazaphosphorines, used in the treatment of certain leukemias, lymphomas and solid tumors, result in significant nephrotoxicity [8-10]. Such off-target toxicity is dose-limiting, thus reducing efficacy.
Alkylating agents represent one class of commonly-utilized chemotherapeutic agents that generate numerous types of alkylated DNA base lesions, including O6-alkylguanine (O6alkG), 7-alkylguanine (7alkG) and 3alkyladenine (3alkA). The induction of cell death in rapidly-dividing cells by these toxic DNA lesions underlies the effectiveness of alkylators as cancer chemotherapeutic agents. The base excision repair (BER) pathway repairs many alkylated DNA base lesions [11]. BER is initiated by the recognition and excision of alkylated DNA base lesions by the alkyladenine DNA glycosylase, Aag (a.k.a. Mpg). After base excision, an AP endonuclease (Ape1) hydrolyzes the phosphodiester backbone at the abasic site, generating a single-stranded DNA break (SSB) with 3’OH and 5’deoxyribose-5-phosphate (5’dRP) termini. Poly(ADP-ribose) polymerase-1 (Parp1) acts as a SSB sensor, and, upon binding a SSB, is activated to catalyze the addition of long polymers of ADP-ribose to itself and several other proteins. The poly(ADP-ribose) chains are thought to recruit downstream BER enzymes: DNA polymerase β (Pol β) that removes the 5’dRP terminus and extends the missing nucleotide from the 3’OH, and either DNA Ligase I or the Xrcc1/LigaseIIIα complex that seal the remaining nicked DNA to complete BER.
Although BER can repair DNA alkylation damage, under certain conditions the initiation of BER can generate toxic repair intermediates that cause damage to healthy tissues. Recently, using mouse genetic models, we demonstrated the importance of both Aag and Parp1 in modulating in vivo alkylation sensitivity. Modest increases in Aag activity in a transgenic mouse model (AagTg mice) increased susceptibility to the alkylating agent, methyl methanesulfonate (MMS), for both whole-animal survival and tissue damage in a specific subset of tissues [12, 13]. The Aag-mediated alkylation sensitivity of these tissues, for both wild-type (WT) and AagTg mice, is entirely Parp1-dependent, being wholly prevented by Parp1 deficiency. Here, we show that although Parp1 deficiency protects against Aag-dependent, MMS-mediated tissue degeneration in the pancreas, spleen, retina, and cerebellum [12], Parp1 deficiency is surprisingly unable to protect against MMS-mediated whole-animal lethality. In fact, AagTg/Parp1-/- mice exhibit greater morbidity and lethality following MMS treatment as compared to AagTg mice. Strikingly, following MMS treatment, AagTg/Parp1-/- mice exhibit severe glomerular damage, suggesting a novel role for Aag and Parp1 in modulating the renal toxicity of alkylating agents.
Numerous animal models of acquired podocyte diseases have been described (reviewed in [14, 15]). MMS-treated AagTg/Parp1-/- mice represent a new model of glomerular disease and may provide insight into many types of human glomerular diseases. Further, the induction of nephrotoxicity by MMS is novel and the importance of Parp1 and Aag in modulating responses is especially relevant, given the recent surge in the use of PARP inhibitors for multiple clinical indications and the wide range of AAG activity in the human population [12, 16, 17].
RESULTS
AagTg and Parp1-/- alleles display synthetic lethality for alkylation-induced animal toxicity
AagTg mice, expressing higher than WT levels of Aag, display increased sensitivity to MMS toxicity compared to WT mice, at the whole-animal level and in numerous tissues including the cerebellum, retina, and pancreas, but not kidney [12, 13]. However, Parp1 deficiency completely suppresses MMS-mediated damage to these tissues in both WT and AagTg mice [12]. To determine whether Parp1 deficiency also protects against whole-body alkylation sensitivity in AagTg mice, we determined the LD50 of the alkylating agent MMS in WT, Parp1-/-, AagTg, and AagTg/Parp1-/- mice. Surprisingly, although alkylation-induced damage in numerous tissues is suppressed in AagTg/Parp1-/- mice [12], whole-animal toxicity is not suppressed; rather, the animals are more susceptible to MMS-induced whole-animal lethality. The MMS LD50 for AagTg mice is 80 mg/kg as we previously reported [12], and 66 mg/kg for AagTg/Parp1-/- mice (Table 1). Parp1-/- mice exhibit the same whole-body sensitivity to MMS as WT (LD50 200 mg/kg). Remarkably, during a 30-day (d) survival experiment post-MMS treatment (75 mg/kg, a dose slightly below the LD50 of AagTg mice), we observed that AagTg/Parp1-/- mice began to lose significant body weight (Figure 1A) and exhibited signs of severe disease, including lethargy, a hunched posture, and cachexia (losing ~20% of initial BW) by 15d post-MMS treatment; all AagTg/Parp1-/- mice succumb within 30d (Figure 1B-1C).
Table 1: Approximate LD50 of MMS in mutant mice
Approximate LD50# |
|
Mouse Strain |
MMS |
WT |
200 mg/kg |
Parp1 -/- |
200 mg/kg |
AagTg |
80 mg/kg |
AagTg/Parp1-/- |
66 mg/kg |
# as determined by Deichmann and LeBlanc method (1943). MMS, methyl methanesulfonate
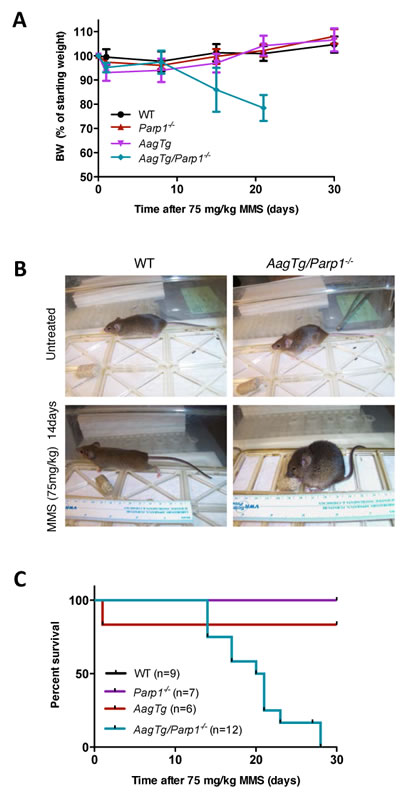
Figure 1: Parp1 deficiency results in increased overall whole-body sensitivity to MMS toxicity. A. Body weight, as percentage of initial body weight following MMS treatment (75 mg/kg), is illustrated for WT (n = 17), Parp1-/- (n = 9), AagTg (n = 19), and AagTg/Parp1-/- (n = 22) mice at different time points. Representative data from two experiments are shown. B. Representative pictures of mouse body condition 14 days post-MMS treatment (75 mg/kg). C. Kaplan Meier survival curves are shown for WT (n = 9), Parp1-/- (n = 7), AagTg (n = 6), and AagTg/Parp1-/- (n = 12) mice following treatment with MMS (75 mg/kg).
AagTg mice combined with either the Parp1-/- allele or Parp inhibition are susceptible to MMS-mediated renal toxicity
To investigate causa mortis in AagTg/Parp1-/- mice, we examined a panel of tissues at gross and histological levels 14d post-MMS. Remarkably, only the kidneys displayed alkylation-induced pathology. MMS-treated AagTg/Parp1-/- kidneys were smaller and paler compared to untreated and MMS-treated WT mice (Figure 2A). Accordingly, severe histological changes were observed. Under low magnification, we observed large, pink, proteinaceous casts that resulted in dilation of the kidney tubules (Figure 2B). High magnification revealed severe glomerular abnormalities, including vacuolation and loss of the Bowman’s space (Figure 2B). Periodic acid-Schiff (PAS) staining shows a glomerular structure severely disturbed in AagTg/Parp1-/- mice post-MMS, as evident by vacuolation and glomerular tuft collapse (Supplemental Figure 1). No evidence of kidney disease was seen in MMS-treated WT, AagTg (Figure 2B and Supplemental Figure 1) or Parp1-/- (not shown) mice. Moreover, no other tissues from MMS-treated AagTg/Parp1-/- mice revealed histological abnormalities (not shown) at 14d. AagTg/Parp1-/- kidneys stained with PAS, toludine blue and phosphotungstic acid-hematoxilin (PTAH) 14d post-MMS showed no evidence of abnormal glomerular cellular accumulations, intravascular fibrin thrombi, matrix deposits, or significant fibrosis (Supplemental Figure 2).
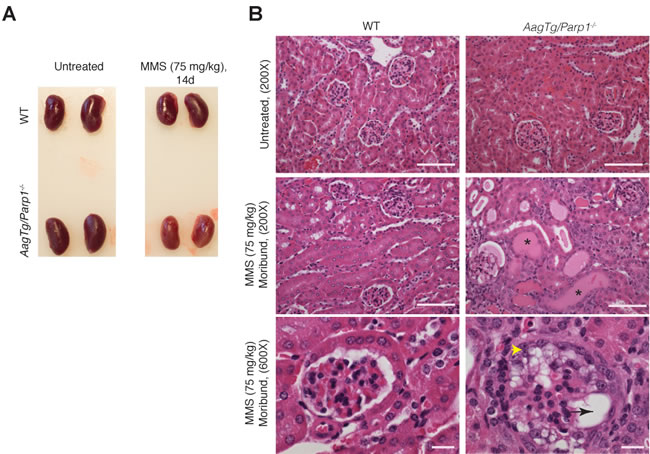
Figure 2: AagTg/Parp1-/- mice exhibit MMS-mediated kidney damage. A. Representative images of kidneys following MMS treatment. Kidneys from a moribund AagTg/Parp1-/- mouse and a WT mouse 14 days post-MMS treatment (75 mg/kg) are shown. B. H&E analysis of kidneys from WT, AagTg, Parp1-/-, and AagTg/Parp1-/- mice 14 days following MMS treatment (75 mg/kg). Magnification is 200X (scale bar 50 µm) and 600x (scale bar 10 µm); asterisks indicate proteinaceous casts, the black arrow indicates vacuolation, and yellow arrowhead indicates parietal and visceral epithelial hypertrophy/hypercellularity with vacuolation, tuft adhesions, capillary collapse and crescent formation.
AagTg mice treated with ABT-888 (a.k.a. Veliparib), a clinically-relevant PARP inhibitor, exhibit MMS-mediated renal toxicity similar to AagTg/Parp1-/- mice (Figure 3) indicating that Parp inhibition was sufficient to sensitize AagTg mouse kidneys to MMS. However, renal toxicity was reduced in MMS/ABT-888-treated AagTg mice compared to MMS-treated AagTg/Parp1-/- mice, with affected glomeruli and protein casts being less prevalent. Consistent with the reduced severity of the kidney phenotype, MMS/ABT-888-treated AagTg mice exhibited lower whole-body sensitivity compared to AagTg/Parp1-/- mice; at 20d post-MMS treatment, no AagTg mice were moribund and >80% of the AagTg mice showed no sign of distress through 30d post-MMS (not shown).
Having shown that AagTg mice combined with either the Parp1-/- allele or Parp inhibition are susceptible to MMS-mediated renal toxicity, we determined whether this toxicity correlates with the accumulation of BER intermediates and lack of Parp activity. The Aag glycosylase generates potentially toxic abasic (AP) sites that can block replication and transcription [18, 19], and their cleavage by AP endonuclease generates highly toxic DNA single-strand breaks [20]; indeed, until the final ligation step of BER, these toxic lesions are present in DNA. Thus, if BER is initiated under conditions where the subsequent processing of BER intermediates is limiting, i.e. by Parp deficiency, this can result in cell death and tissue damage; under these circumstances cells are said to have an imbalanced BER pathway [11]. Indeed, at 3 hours post-MMS treatment, AagTg kidney DNA contained twice as many AP sites as WT kidney DNA (Supplemental Figure 3A). Consistent with these findings, at 6 hours post-MMS treatment, Parp activity was highly increased in AagTg kidneys compared to WT kidneys (Supplemental Figure 3B). In both WT and AagTg kidneys, Parp activity peaks at 24 hours post-MMS treatment and returns to basal levels within 48 hours post-MMS treatment. Conversely, although AagTg/Parp1-/- kidney DNA had high levels of AP sites similarly to AagTg kidney DNA at 3 hours post-MMS treatment (Supplemental Figure 3A), no increase in Parp activity was observed in AagTg/Parp1-/- kidneys at any time point (Supplemental Figure 3B). These findings support the hypothesis that an imbalanced BER pathway can be detrimental for the kidney.
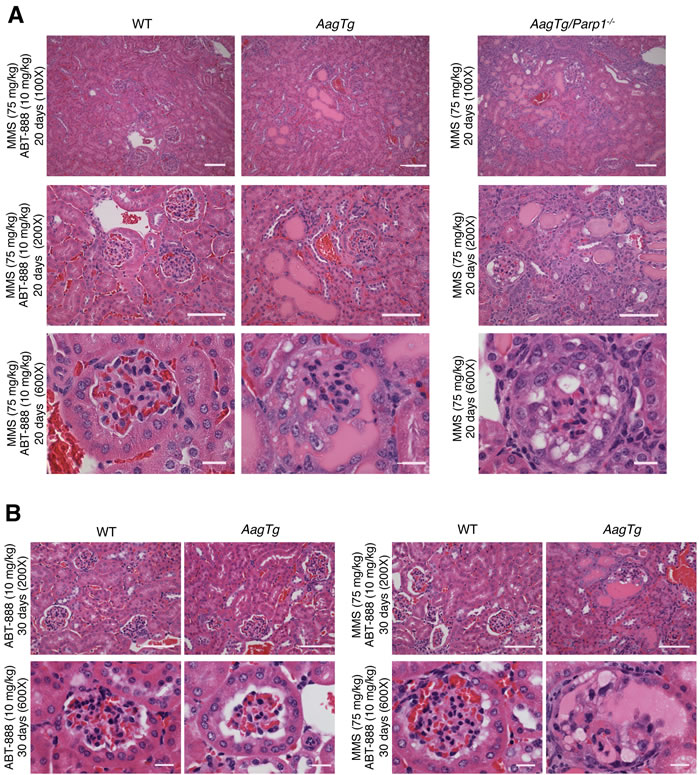
Figure 3: AagTg mice treated with a Parp inhibitor exhibit MMS-mediated renal toxicity similar to AagTg/Parp1-/- mice. H&E stained images of kidneys from WT, AagTg, and AagTg/Parp1-/- mice 20 days (A) and 30 days (B) following MMS (75 mg/kg) and/or ABT-888 (10 mg/kg) treatment as indicated. ABT-888 was administered 1 hour prior and 5 days post-MMS treatment. Magnification is 100X, 200X (scale bar 50 µm) and 600x (scale bar 10 µm); MMS- and ABT-888-treated AagTg mice show islands of pink proteinaceous casts, dilation of the kidney tubules, and few glomeruli with severe abnormalities (including vacuolation and the loss of Bowman’s space) as seen in AagTg/Parp1-/- mice post-MMS treatment.
Progression of MMS-mediated renal toxicity in AagTg/Parp1-/- mice
We examined the kinetics of MMS-induced kidney damage by monitoring kidney toxicity at 3, 5, 7 and 14d post-MMS. We assessed the glomerulus, renal tubules and Bowman’s capsule following histopathological criteria listed in Supplemental Table 1. No histopathological abnormalities were observed at any time point in WT mice and in AagTg/Parp1-/- mice at 3d or 5d post-MMS (Figure 4A-4C). However, by 7d we observe abnormalities in AagTg/Parp1-/- kidneys. One obvious change within the renal tubules at 7d post-MMS is tubule dilation and presence of proteinaceous casts (Figure 4C). By 14d there is also evidence of reactive changes in the tubules, including mild degeneration and hyperplasia (Figure 4C). Consistent with reactive regeneration, epithelial mitotic figures are also observed (Supplemental Figure 4). The glomerulus exhibits histological abnormalities as early as 7d post-MMS, including vacuolation of podocytes or parietal cells and capillary collapse of the lumen (Figure 4D). By 14d, glomeruli in AagTg/Parp1-/- mice exhibit significant increases in the histological scores of podocyte or parietal cell hyperplasia (p = 0.0005), vacuolation (p = 0.001) and capillary lumen collapse (p < 0.05). At 14d post-MMS, additional glomerular pathological abnormalities appear, including crescent formation (Figure 2B and 4A), a phenomenon commonly observed in crescentic glomerulonephritis in both humans and mice [21-23].
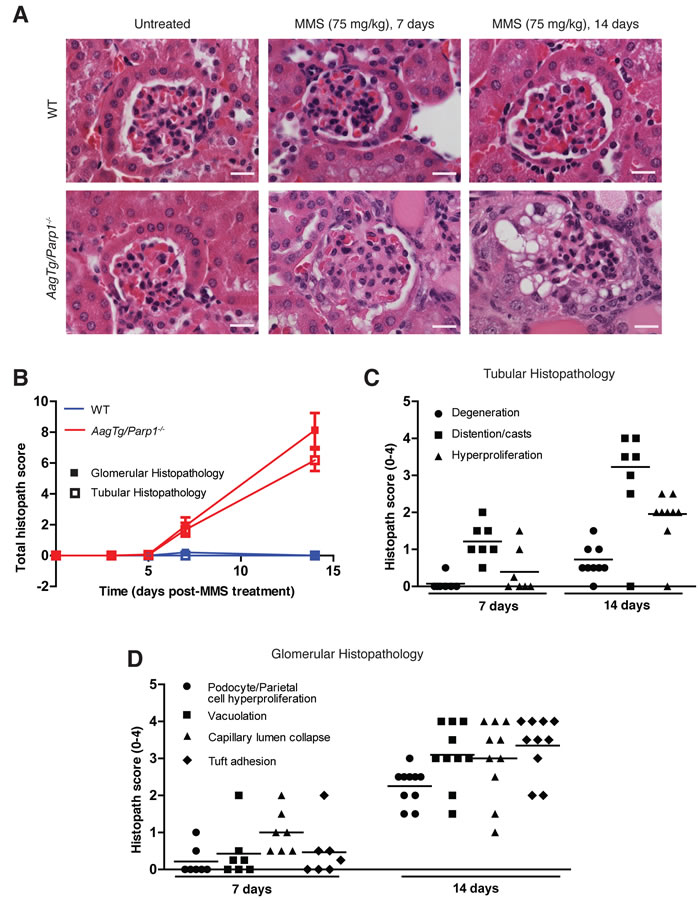
Figure 4: Kinetics of kidney damage in AagTg/Parp1-/-mice following MMS treatment. A. H&E-stained images of kidneys from WT and AagTg/Parp1-/- mice under untreated conditions and 7 and 14 days following MMS treatment (75 mg/kg). Magnification is 600X (scale bar 10 µm). B. Total histopathology scores for glomerular damage (podocyte/parietal cell hyperplasia/hypertrophy, vacuolation, capillary lumen collapse, tuft adhesions and/or sclerosis) and tubular damage (degeneration, cast formation, hyperplasia) is shown at five time points, post MMS treatment (75 mg/kg); untreated (WT, n = 5 and AagTg/Parp1-/-, n = 7), 3 days (WT, n = 3 and AagTg/Parp1-/-, n = 2), 5 days (WT, n = 7 and AagTg/Parp1-/-, n = 10), 7 days (WT, n = 5 and AagTg/Parp1-/-, n = 7 and 14 days (WT, n = 10 and AagTg/Parp1-/-, n = 11). Histopathological scores for C. renal tubule criteria and D. glomerular criteria.
AagTg/Parp1-/- mice exhibit clinical markers of kidney disease
We next investigated whether kidney abnormalities observed at the histological level were accompanied by clinical markers of disease. Serum analytes were measured before treatment, 14d post-MMS, and when the mice exhibited morbidity post-MMS. We observe a trend toward higher serum levels of blood urine nitrogen (BUN) in AagTg/Parp1-/- mice 14d post-MMS compared to WT mice (p = 0.157) (Figure 5A); this trend became significant in moribund AagTg/Parp1-/- mice (p < 0.005) (Figure 5A). Levels of blood creatinine, another marker of kidney damage, did not change significantly post-MMS treatment (Figure 5B).
Serum albumin levels, a marker of glomerular function, displayed dramatic decreases following MMS in AagTg/Parp1-/- mice compared to treated WT mice or untreated AagTg/Parp1-/- mice (Figure 5C). In addition, we observed high levels of a ~65 kDa protein, consistent with the size of albumin, in the AagTg/Parp1-/- mouse urine 7d post-MMS (Figure 5D). Together, these data indicate that albumin is not properly maintained in the serum, but is likely accumulating in the kidney tubules as proteinaceous casts, and ultimately leaking into the urine of MMS-treated AagTg/Parp1-/- mice. The albumin leakage in the urine 7d post-MMS is consistent with histopathological changes within the glomerulus observed at the same time point (Figure 5D). These features indicate impaired glomerular function induced by MMS.
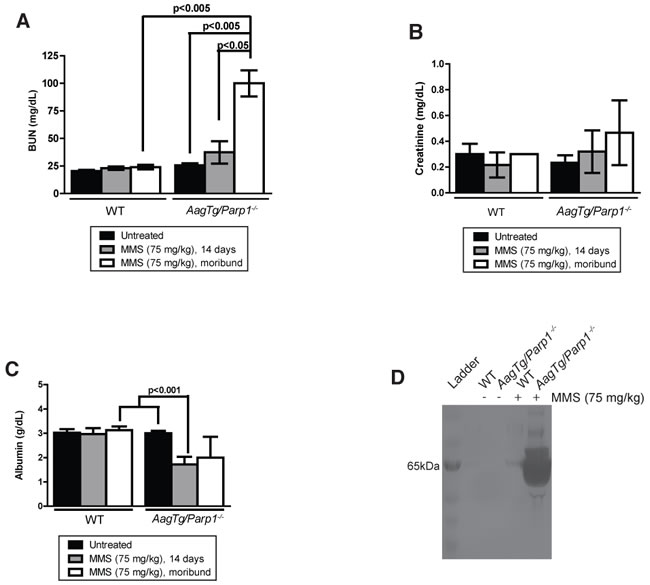
Figure 5: AagTg/Parp1-/- mice exhibit clinical signs of kidney damage. Serum levels of A. blood urea nitrogen (BUN), B. creatinine, and C. albumin were measured in untreated (n = 3-4), 14 days post-MMS treatment (75 mg/kg) (n = 5-6), and when mice exhibit morbidity (n = 3). For the moribund time point, a control WT mouse was assayed with each AagTg/Parp1-/- mouse. D. Silver stain visualization of protein content in urine of WT and AagTg/Parp1-/- in untreated conditions and 7 days following MMS treatment (75 mg/kg).
AagTg/Parp1-/- mice exhibit MMS-induced podocyte injury
Given the apparent loss of glomerular filtration, we examined whether the AagTg/Parp1-/- mice displayed podocyte injury post-MMS by immunostaining for Wilm’s tumor 1 (WT1) protein, a podocyte marker. MMS-treated AagTg/Parp1-/- mouse kidneys exhibited a decreased number of WT1-stained cells. No significant change was observed in MMS-treated WT mice (Figure 6A-6B). Furthermore, we investigated a potential alteration in the slit diaphragm by immunostaining for Podocin. MMS-treated AagTg/Parp1-/- mice exhibit diminished staining of Podocin, consistent with reduced glomerular filtration (Figure 6C).
Electron-microscopy (Figure 6D) revealed that WT mice (untreated and14d post-MMS) had normal glomerular tuft architecture comprised of well-organized capillary loops, Bowman’s capsule and flat parietal epithelial cells (PECs). Podocytes looked healthy with numerous well-organized foot processes. Untreated AagTg/Parp1-/- mouse kidneys appeared similar to the WT controls in ultrastructural morphology of glomerular tufts. However, MMS-treated AagTg/Parp1-/- mouse kidneys (14d) showed ultrastructural alterations consistent with the main histological alterations observed in the podocytes and PECs. As shown in Figure 6D, PECs and podocytes were markedly hypertrophied by the presence of multiple intracytoplasmic medium-to-large-sized membrane-bound vacuoles. These vacuoles were mostly empty or occasionally contained pale to electron-dense irregular granular to amorphous material. The capillary tufts were distorted and compressed of numerous large distinct vacuoles within the podocyte foot processes. Extensive flattening and alteration of podocyte foot processes was observed. There was also a discernible reduction in the area of capillary loop and fusion between the large vacuolated PECs and podocytes at the urinary pole, resulting in loss of urinary space. These observations confirm that AagTg/Parp1-/- mice exhibit severe morphological and functional podocyte damage post-MMS.
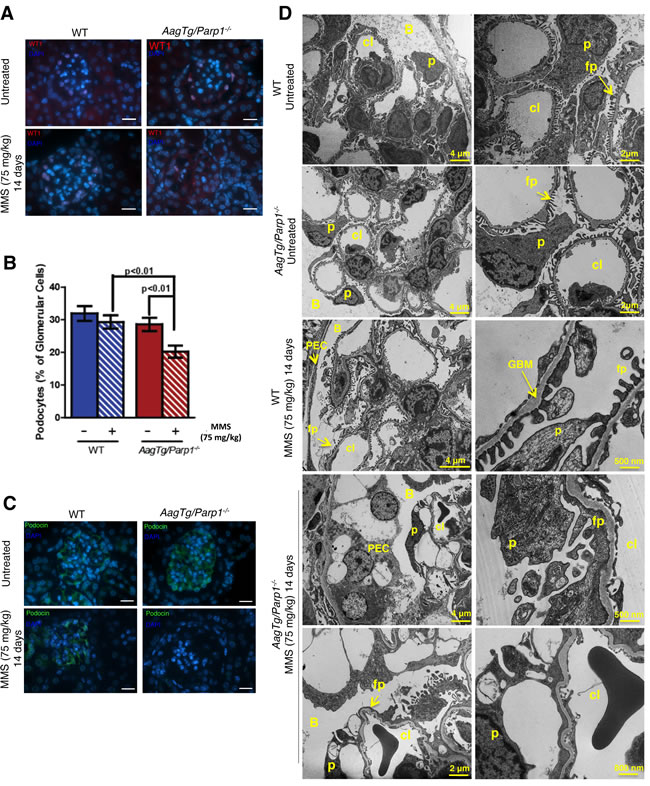
Figure 6: Podocyte damage is observed following MMS treatment in AagTg/Parp1-/- mice. A. Immunofluorescence staining of WT1-positive podocytes in WT and AagTg/Parp1-/- mice, untreated and 14 days following MMS treatment (75 mg/kg). A representative image of n = 3 mice is shown. Magnification is 600X (scale bar 10 µm). B. Quantitation of podocytes (the % of cells stained with both WT1 and DAPI/total number of DAPI positive cells in the glomerulus) is shown. Eight glomeruli were counted on each section from n = 3-4 mice/condition. C. Immunofluorescence staining of Podocin in WT and AagTg/Parp1-/- mice in untreated and 14 days following MMS treatment (75 mg/kg). Magnification is 600X (scale bar 10 µm). D. Transmission electron microscopy (TEM) of WT and AagTg/Parp1-/- mice, untreated and 14 days following MMS treatment (75 mg/kg). P, podocytes; fp, foot processes; cl, capillary lumen; B, Bowman’s capsule; GBM, glomerular basement membrane; PEC, parietal epithelial cell.
Female AagTg/Parp1-/- mice are protected from MMS-induced nephrotoxicity
An unexpected sexual dimorphism became apparent in MMS-treated female AagTg/Parp1-/- mice. MMS-induced whole-animal toxicity was reduced in AagTg/Parp1-/- female mice. The MMS LD50 for AagTg/Parp1-/- female mice is 80 mg/kg, whereas that for AagTg/Parp1-/- male mice is 66 mg/kg. Furthermore, in contrast with the morbidity observed in male AagTg/Parp1-/- mice, female AagTg/Parp1-/- mice exhibited no body weight decrease and survived 30d post-MMS (75 mg/kg, Figure 7A-7B). Histopathological analysis confirmed complete protection against kidney damage in MMS-treated female versus male AagTg/Parp1-/- mice (Figure 7C-7D). The renoprotective roles of estrogen are well-established in both mice [24, 25] and humans [26, 27]; our data suggest that estrogen may also be protective against alkylation-induced nephrotoxicity in AagTg/Parp1-/- mice. Indeed, chronic 17b-estradiol treatment (E2 pellets) completely protected 6 out of 8 male mice against MMS-induced kidney damage (Supplemental Figure 5). The remaining 2 mice showed only a mild pathology with few alterations of tubules and few affected glomeruli. Moreover, MMS-mediated nephrotoxicity in males is not a consequence of the genetic background or strain, as AagTg/Parp1-/- mice on a pure 129S background exhibit kidney functional abnormalities similar to that of AagTg/Parp1-/- mice on a C57Bl/6J:129S mixed background (Supplemental Figure 6). This finding is especially relevant because genetic background has previously been shown to modify susceptibility to glomerular disorders [28, 29].
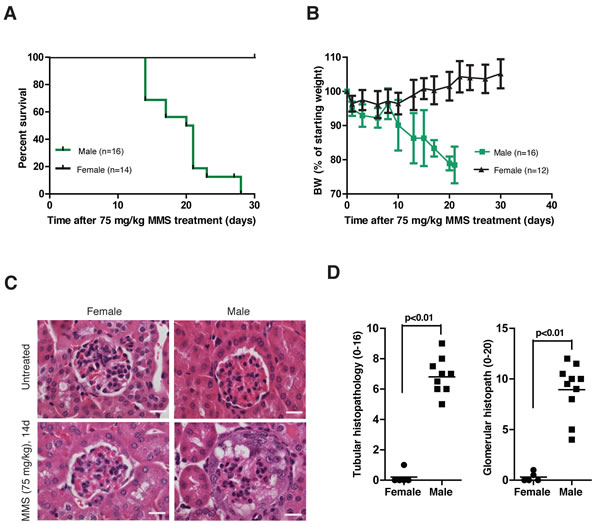
Figure 7: Female AagTg/Parp1-/- mice are protected from MMS-mediated kidney damage. A. Kaplan Meier survival curves are shown for female (n = 14) and male (n = 16) AagTg/Parp1-/- mice following treatment with MMS (75 mg/kg). B. BW, as percent of initial BW, is shown for female AagTg/Parp1-/- (n = 14) and male AagTg/Parp1-/- (n = 16) mice following treatment with MMS (75 mg/kg). C. H&E-stained images from kidneys are shown for female and male AagTg/Parp1-/- mice 14 days following MMS treatment (75 mg/kg). Magnification is 600x (scale bar 10 µm). D. Total histopathology scores for glomerular damage (podocyte/parietal cell hyperplasia/hypertrophy, vacuolation, capillary lumen collapse, tuft adhesions/sclerosis) and tubular damage (degeneration, cast formation, hyperplasia) is shown for female and male mice 14 days post-MMS treatment (75 mg/kg). Scores in a AagTg/ Parp1-/- male is also presented in Figure 4B-4D.
DISCUSSION
Although protected from MMS-induced toxicity in a variety of tissues, AagTg/Parp1-/- mice eventually succumb to whole-animal lethality at lower MMS doses in comparison to AagTg mice. This increased lethality was dependent on imbalanced BER, caused by increased Aag levels, since the MMS LD50 in Parp1-/- mice expressing normal Aag levels is equivalent to that in WT mice. Interestingly, when BER is imbalanced by increased Aag activity, there are obvious tissue-specific differences with respect to whether Parp1 deficiency suppresses or exacerbates toxicity. Parp1 deficiency protects AagTg mice against MMS-induced tissue damage in the cerebellum, pancreas, spleen and retina [12], but enhances MMS-induced kidney damage. We examined a multitude of tissues in AagTg/Parp1-/- mice post-MMS and only found histological abnormalities in the kidney. It is possible that with time, additional tissues could display similar damage and that the rapid development of kidney disease prevents observation of such damage. Controlling Aag expression in a tissue-specific manner will help to determine whether other tissues exhibit MMS-mediated toxicity in the absence of Parp1 at longer time points. Neither increased Aag activity, nor Parp1 deficiency alone resulted in susceptibility to kidney damage, but rather the combination of both gave rise to this severe kidney phenotype post-MMS treatment. We found that AagTg/Parp1-/- mice as well as AagTg mice generate more BER intermediates, i.e. AP sites, than WT mice post-MMS treatment. However, in AagTg mice, the higher accumulation of AP sites is compensated by higher levels of Parp activity compared to WT. In both, WT and AagTg mice, Parp activity returns to basal levels within 48 hours when all lesions have likely been repaired. Unlike AagTg mice, AagTg/Parp1-/- mice do not show any Parp activity, suggesting that Parp deficiency is a limiting step in subsequent processing of BER intermediates and that the accumulation of BER intermediates, due to increased Aag activity and Parp deficiency, might be the cause of kidney toxicity. Our results are especially relevant given the fact that AAG levels can vary >10-fold in the human population (at least in lymphocytes) [12, 16, 17] and given the recent surge in the use of PARP inhibitors in combination with chemotherapeutic agents. As nephrotoxicity can be the rate-limiting side effect in a subset of chemotherapeutic agents, these findings may have important clinical relevance [30].
In contrast to our finding that MMS induces nephrotoxicity in Parp1-/- and Parp-inhibited AagTg mice, several other models of kidney damage have demonstrated protection under conditions of genetic or pharmacologic Parp depletion. Parp1 depletion protects against nephrotoxicity induced by the chemotherapeutic agent, cisplatin [31, 32], diabetic nephropathy [33, 34] and the necrosis associated with renal ischemia/reperfusion [35, 36]. The MMS-induced nephrotoxicity in AagTg/Parp1-/- mice was unexpected and to our knowledge has not been previously described. This finding suggests that Parp1 deletion or Parp inhibition may not give rise to the same outcomes across all tissues and experimental models, making it increasingly important to consider the clinical consequences of PARP inhibition in cancer treatment.
Parp1 is a multi-functional protein, exhibiting many functions independent of its role in DNA repair [37]. It is possible that a DNA repair-independent Parp1 function contributes to the phenotype described here. For example, Parp1 plays a role in modulating inflammation [38, 39] and although there is no evidence of altered immune cell recruitment to the glomeruli, it remains possible that immune system changes contribute to this severe glomerular phenotype. Indeed, autoimmune disorders, such as lupus nephritis, routinely result in glomerular lesions with some of the characteristics we observe [40].
Strikingly, female AagTg/Parp1-/- mice were completely protected from MMS-mediated nephrotoxicity. Males (in both human and animal models) are more prone to kidney disease [41, 42]. Indeed, epidemiological studies identified male sex as a risk factor for kidney disease [43]. Consistent with this, female mice are protected from ischemic renal injury and diabetic nephropathy, and estrogen provides protection against ischemia-reperfusion renal injury and age-related kidney decline [44-46], but does not protect against cisplatin-mediated nephrotoxicity [47]. Specifically, stimulation of estrogen receptors protects against podocyte damage and death both in vitro and in vivo [48, 49]. Not only is estrogen protective, but testosterone potentiates renal injury [50-53]. Here, we show that estrogen treatment protects male mice from alkylation-induced nephrotoxicity. These findings further support the role of estrogen in protection against kidney disease. However, in addition to sex steroids, sexual dimorphisms in renal morphology and physiology [54, 55] may also contribute to the protection of female AagTg/Parp1-/- mice from MMS-mediated kidney disease. Additionally, it has been described previously that female and male mice exhibit different responses to Parp1 deletion or Parp inhibition in mouse models of endotoxemia and nephrotoxic serum-induced nephritis [56, 57]. These data further underscore the recent push for investigating and understanding sex differences in basic and clinical science [58-61].
The sexual dimorphism in our model may be explained by the interaction of Aag and Parp1 with estrogen receptor-α (ERα) [62, 63]. A direct interaction between Aag and ERα has been described; binding of Aag and ERα causes increased catalytic activity of Aag and decreased ERα-mediated transcription [62]. Parp1 has been shown to bind and PARylate ERα, leading to increased binding of ERα to the estrogen response element in the promoter of target genes, thus promoting ERα-mediated transcription [63]. Together, these findings suggest that a direct interaction between Aag and/or Parp1 with estrogen receptors could lead to changes in DNA repair and/or gene expression, explaining the sexual dimorphism of MMS-mediated nephrotoxicity in mice.
Numerous existing models of acquired podocyte diseases are used to explore human renal diseases, however all models have both advantages and disadvantages [14]. We present here a new model that may provide insight into the mechanisms of podocyte injury and death following treatment with alkylating agents.
MATERIALS AND METHODS
Animals and treatments
Parp1-/- mice were purchased from Jackson Laboratories [64]. Aag transgenic (AagTg) mice were described previously [12, 13, 65]. All experiments were performed in mixed background mice (C57Bl/6J:129S) unless otherwise stated. All animal procedures were performed according the NIH guide for the Care and Use of Laboratory Animals.
Approximate LD50 was determined as in Deichmann and LeBlanc [66]. MMS was injected intraperitoneally. ABT-888 (10 mg/kg, Selleck Chemicals Inc) was administered by oral gavage 1 hour before and 5 days after MMS treatment. For estrogen treatment, 17b-estradiol pellets (E2, 0.1 mg/21 days release, Innovative Research of America) were implanted s.c. in the dorsal neck region of adult male mice. MMS was administered 3 days after implants.
AP sites and PARP activity measurement
DNA was isolated using Roche kit according to the manufacturer’s instructions with precautions taken to avoid overheating of the DNA solution (proteinase K step 10 min at 55 0C). AP sites were determined by using the DNA Damage Quantification Kit (Dojindo) and performed according to the manufacturer’s instructions. Parp activity was determined by using the HT PARP in vivo Pharmacodynamic Assay II (Trevigen) according to the manufacturer’s instructions.
Serum analytes and albumin gel electrophoresis
Whole blood was collected via cardiac puncture and placed in BD microtainer serum separator tubes (VWR). Blood samples were then centrifuged for 5 minutes at 2000 x g to separate serum. Idexx Laboratories, Inc. (North Grafton, MA) performed serum chemistry analysis.
Two μL of urine was denatured and run on a SDS-PAGE gel and proteins were visualized using ProteoSilver Plus Silver Staining Kit (Sigma).
Electron microscopy
Tissue processing and imaging was performed by the W.M. Keck Microscopy Facility at the Whitehead Institute (MIT). Kidney cortex was fixed in 2.5% gluteraldehyde, 3% paraformaldehyde with 5% sucrose in 0.1 M sodium cacodylate buffer (pH 7.4), post-fixed in 1% osmium tetroxidein veronal-acetate buffer, stained in block overnight with 0.5% uranyl acetate in veronal-acetate buffer (pH 6.0) and then embedded in Embed-812 resin. Ultrathin sections were cut from blocks on a Leica Ultracut UCT microtome with a Diatome diamond knife. The sections were examined using a FEI Tecnai Spirit at 80 keV.
Histopathology
Tissues were processed by the Histology Core Facility at the David H. Koch Institute for Integrative Cancer Research (MIT); they were paraffin-embedded, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E), or Periodic Acid-Schiff (PAS). Phosphotungstic acid-haematoxylin (PTAH) staining was performed at the Division of Comparative Medicine (MIT). All H&E stained slides were blindly analyzed by a pathologist (R.T.B). Renal lesions were blindly scored by a pathologist (S.M.) using criteria listed in Supplemental Table 1.
Immunofluorescence
Deparaffinized tissue sections (5 μm) were thermally processed for epitope retrieval using a Prestige Medical 2100-Retriver. Sections were permeabilized in PBS-T (1x PBS + 0.1% Triton X-100, three times for 5 minutes each ), incubated with 1x PBS-T plus 10% goat serum for 1 hour and then stained with either anti-WT1antibody (Santa Cruz) or anti-podocin (Sigma) overnight at 4oC. Nuclear counterstaining was done using DAPI. The number of glomerular DAPI- and WT1-stained cells were counted using ImageJ.
Statistics
Statistical analyses were performed using GraphPad Prism software. Statistical significance was determined using unpaired t-test. Kaplan-Meier survival curves were generated and survival differences determined using the Log-Rank test. Data are represented as mean +/- SEM. A p-value is considered significant if less than 0.05.
ACKNOWLEDGMENTS
We thank the Hope Babette Tang Histology Core at the David H. Koch Institute for Integrative Cancer Research and Nicki Watson from W.M. Keck Microscopy Facility at the Whitehead Institute for technical help.
CONFLICTS OF INTEREST
The authors declare that no conflict of interest exists.
GRANT SUPPORT
This work was supported by NIH grants R01-CA075576, R01-CA055042, R01-CA149261, AG-SS-3046-12, P30-ES02109 and P30-CA014051. L.D.S is an American Cancer Society Research Professor and an Ellison Medical Foundation Senior Scholar.
REFERENCES
1. Büscher AK and Weber S. Educational paper: The podocytopathies. Eur J Pediatr. 2012; 171:1151-1160.
2. Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney international. 2007; 71:1205-1214.
3. Gardenswartz MH, Lerner CW, Seligson GR, Zabetakis PM, Rotterdam H, Tapper ML, Michelis MF and Bruno MS. Renal disease in patients with AIDS: a clinicopathologic study. Clin Nephrol. 1984; 21:197-204.
4. Segelmark M and Hellmark T. Autoimmune kidney diseases. Autoimmunity Reviews. 2010; 9:A366-A371.
5. U.S. Renal Data System, USRDS 2003 Annual Data Report: Atlas of End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2003. Available at (https://www.usrds.org/atlas03.aspx).
6. Brinkkoetter PT, Ising C and Benzing T. The role of the podocyte in albumin filtration. Nat Rev Nephrol. 2013; 9:328-336.
7. Moreno JA, Sanchez-Nino MD, Sanz AB, Lassila M, Holthofer H, Blanco-Colio LM, Egido J, Ruiz-Ortega M and Ortiz A. A slit in podocyte death. Curr Med Chem. 2008; 15:1645-1654.
8. Roth BJ. Ifosfamide in the treatment of bladder cancer. Semin Oncol. 1996; 23:50-55.
9. Loebstein R, Atanackovic G, Bishai R, Wolpin J, Khattak S, Hashemi G, Gobrial M, Baruchel S, Ito S and Koren G. Risk Factors for Long-Term Outcome of Ifosfamide-Induced Nephrotoxicity in Children. The Journal of Clinical Pharmacology. 1999; 39:454-461.
10. Skinner R. Chronic ifosfamide nephrotoxicity in children. Medical and Pediatric Oncology. 2003; 41:190-197.
11. Fu D, Calvo JA and Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012; 12:104-120.
12. Calvo JA, Moroski-Erkul CA, Lake A, Eichinger LW, Shah D, Jhun I, Limsirichai P, Bronson RT, Christiani DC, Meira LB and Samson LD. Aag DNA glycosylase promotes alkylation-induced tissue damage mediated by Parp1. PLoS Genet. 2013; 9:e1003413.
13. Meira LB, Moroski-Erkul CA, Green SL, Calvo JA, Bronson RT, Shah D and Samson LD. Aag-initiated base excision repair drives alkylation-induced retinal degeneration in mice. Proc Natl Acad Sci U S A. 2009; 106:888-893.
14. Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, Durvasula RV, Hauser PV, Kowalewska J, Krofft RD, Logar CM, Marshall CB, Ohse T and Shankland SJ. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009; 296:F213-229.
15. Leeuwis JW, Nguyen TQ, Dendooven A, Kok RJ and Goldschmeding R. Targeting podocyte-associated diseases. Adv Drug Deliv Rev. 2010; 62:1325-1336.
16. Crosbie PA, Watson AJ, Agius R, Barber PV, Margison GP and Povey AC. Elevated N3-methylpurine-DNA glycosylase DNA repair activity is associated with lung cancer. Mutation Research. 2012; 732:43-46.
17. Leitner-Dagan Y, Sevilya Z, Pinchev M, Kremer R, Elinger D, Rennert HS, Schechtman E, Freedman L, Rennert G, Livneh Z and Paz-Elizur T. Enzymatic MPG DNA repair assays for two different oxidative DNA lesions reveal associations with increased lung cancer risk. Carcinogenesis. 2014; 35:2763-2770.
18. Kothandapani A, Dangeti VSMN, Brown AR, Banze LA, Wang X-H, Sobol RW and Patrick SM. Novel Role of Base Excision Repair in Mediating Cisplatin Cytotoxicity. Journal of Biological Chemistry. 2011; 286:14564-14574.
19. Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK and Wilson SH. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000; 405:807-810.
20. Liu Y, Prasad R, Beard WA, Kedar PS, Hou EW, Shock DD and Wilson SH. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J Biol Chem. 2007; 282:13532-13541.
21. Tarzi RM, Sharp PE, McDaid JP, Fossati-Jimack L, Herbert PE, Pusey CD, Cook HT and Warrens AN. Mice with defective Fas ligand are protected from crescentic glomerulonephritis. Kidney Int. 2012; 81:170-178.
22. Hamano Y, Abe M, Matsuoka S, Zhang D, Kondo Y, Kagami Y, Ishigami A, Maruyama N, Tsuruta Y, Yumura W and Suzuki K. Susceptibility QTLs for pathogenic leukocytosis in SCG/Kj mice, a spontaneously occurring crescentic glomerulonephritis and vasculitis model. Clinical & Experimental Immunology. 2014:n/a-n/a.
23. Moroni G and Ponticelli C. Rapidly progressive crescentic glomerulonephritis: Early treatment is a must. Autoimmunity Reviews. 2014; 13:723-729.
24. Chin M, Isono M, Isshiki K, Araki S, Sugimoto T, Guo B, Sato H, Haneda M, Kashiwagi A and Koya D. Estrogen and raloxifene, a selective estrogen receptor modulator, ameliorate renal damage in db/db mice. The American Journal of Pathology. 2005; 166:1629-1636.
25. Tanaka R, Tsutsui H, Ohkita M, Takaoka M, Yukimura T and Matsumura Y. Sex differences in ischemia/reperfusion-induced acute kidney injury are dependent on the renal sympathetic nervous system. Eur J Pharmacol. 2013; 714:397-404.
26. Neugarten J, Acharya A and Silbiger SR. Effect of gender on the progression of nondiabetic renal disease: a meta-analysis. Journal of the American Society of Nephrology : JASN. 2000; 11:319-329.
27. (2000). U.S. Renal Data System:USRDS 2000Annual data Report.NIDDK, The National Institute of Health:Bethesda, MD.
28. Bygrave AE, Rose KL, Cortes-Hernandez J, Warren J, Rigby RJ, Cook HT, Walport MJ, Vyse TJ and Botto M. Spontaneous autoimmunity in 129 and C57BL/6 mice-implications for autoimmunity described in gene-targeted mice. PLoS Biol. 2004; 2:E243.
29. Uchio-Yamada K, Sawada K, Tamura K, Katayama S, Monobe Y, Yamamoto Y, Ogura A and Manabe N. Tenc1-deficient mice develop glomerular disease in a strain-specific manner. Nephron Exp Nephrol. 2013; 123:22-33.
30. Perazella MA and Moeckel GW. Nephrotoxicity From Chemotherapeutic Agents: Clinical Manifestations, Pathobiology, and Prevention/Therapy. Seminars in Nephrology. 2010; 30:570-581.
31. Mukhopadhyay P, Horvath B, Kechrid M, Tanchian G, Rajesh M, Naura AS, Boulares AH and Pacher P. Poly(ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic Biol Med. 2011; 51:1774-1788.
32. Kim J, Long KE, Tang K and Padanilam BJ. Poly(ADP-ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int. 2012; 82:193-203.
33. Shevalye H, Maksimchyk Y, Watcho P and Obrosova IG. Poly(ADP-ribose) polymerase-1 (PARP-1) gene deficiency alleviates diabetic kidney disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2010; 1802:1020-1027.
34. Szabó C, Biser A, Benk R, Böttinger E and Suszták K. Poly(ADP-Ribose) Polymerase Inhibitors Ameliorate Nephropathy of Type 2 Diabetic Leprdb/db Mice. Diabetes. 2006; 55:3004-3012.
35. Martin DR, Lewington AJP, Hammerman MR and Padanilam BJ. Inhibition of poly(ADP-ribose) polymerase attenuates ischemic renal injury in rats. Journal Article. 2000; 279:R1834-R1840.
36. Zheng J, Devalaraja-Narashimha K, Singaravelu K and Padanilam BJ. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. American Journal of Physiology - Renal Physiolog. 2005; 288:F387-F398.
37. Weaver AN and Yang ES. Beyond DNA repair: additional functions of PARP-1 in cancer. Frontiers in Oncology. 2013; 3.
38. Rosado MM, Bennici E, Novelli F and Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology. 2013; 139:428-437.
39. Bai P and Virág L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Letters. 2012; 586:3771-3777.
40. Schwartz MM. The Pathology of Lupus Nephritis. Seminars in Nephrology. 2007; 27:22-34.
41. Neugarten J and Golestaneh L. Gender and the Prevalence and Progression of Renal Disease. Advances in Chronic Kidney Disease. 2013; 20:390-395.
42. Neugarten J, Acharya A and Silbiger SR. Effect of gender on the progression of nondiabetic renal disease: a meta-analysis. J Am Soc Nephrol. 2000; 11:319-329.
43. Yanes LL, Sartori-Valinotti JC and Reckelhoff JF. Sex steroids and renal disease: lessons from animal studies. Hypertension. 2008; 51:976-981.
44. Hutchens MP, Nakano T, Kosaka Y, Dunlap J, Zhang W, Herson PS, Murphy SJ, Anderson S and Hurn PD. Estrogen is renoprotective via a nonreceptor-dependent mechanism after cardiac arrest in vivo. Anesthesiology. 2010; 112:395-405.
45. Tanaka R, Tsutsui H, Kobuchi S, Sugiura T, Yamagata M, Ohkita M, Takaoka M, Yukimura T and Matsumura Y. Protective effect of 17beta-estradiol on ischemic acute kidney injury through the renal sympathetic nervous system. Eur J Pharmacol. 2012; 683:270-275.
46. Inada A, Arai H, Nagai K, Miyazaki J, Yamada Y, Seino Y and Fukatsu A. Gender difference in ICER Igamma transgenic diabetic mouse. Biosci Biotechnol Biochem. 2007; 71:1920-1926.
47. Pezeshki Z, Nematbakhsh M, Nasri H, Talebi A, Pilehvarian AA, Safari T, Eshraghi-Jazi F, Haghighi M and Ashrafi F. Evidence against protective role of sex hormone estrogen in Cisplatin-induced nephrotoxicity in ovarectomized rat model. Toxicol Int. 2013; 20:43-47.
48. Kummer S, Jeruschke S, Wegerich LV, Peters A, Lehmann P, Seibt A, Mueller F, Koleganova N, Halbenz E, Schmitt CP, Bettendorf M, Mayatepek E, Gross-Weissmann ML and Oh J. Estrogen receptor alpha expression in podocytes mediates protection against apoptosis in-vitro and in-vivo. PLoS One. 2011; 6:e27457.
49. Catanuto P, Fornoni A, Pereira-Simon S, Wu F, Burnstein KL, Xia X, Conti F, Lenzi A and Elliot S. In vivo 17beta-estradiol treatment contributes to podocyte actin stabilization in female db/db mice. Endocrinology. 2012; 153:5888-5895.
50. Verzola D, Gandolfo MT, Salvatore F, Villaggio B, Gianiorio F, Traverso P, Deferrari G and Garibotto G. Testosterone promotes apoptotic damage in human renal tubular cells. Kidney Int. 2004; 65:1252-1261.
51. Long DA, Kolatsi-Joannou M, Price KL, Dessapt-Baradez C, Huang JL, Papakrivopoulou E, Hubank M, Korstanje R, Gnudi L and Woolf AS. Albuminuria is associated with too few glomeruli and too much testosterone. Kidney Int. 2013; 83:1118-1129.
52. Hodeify R, Megyesi J, Tarcsafalvi A, Mustafa HI, Hti Lar Seng NS and Price PM. Gender differences control the susceptibility to ER stress-induced acute kidney injury. American Journal of Physiology - Renal Physiology. 2013; 304:F875-F882.
53. Doublier S, Lupia E, Catanuto P, Periera-Simon S, Xia X, Korach K, Berho M, Elliot SJ and Karl M. Testosterone and 17beta-estradiol have opposite effects on podocyte apoptosis that precedes glomerulosclerosis in female estrogen receptor knockout mice. Kidney Int. 2011; 79:404-413.
54. Sabolic I, Asif AR, Budach WE, Wanke C, Bahn A and Burckhardt G. Gender differences in kidney function. Pflugers Arch. 2007; 455:397-429.
55. Yabuki A, Suzuki S, Matsumoto M and Nishinakagawa H. Sexual dimorphism of proximal straight tubular cells in mouse kidney. Anat Rec. 1999; 255:316-323.
56. Jog NR and Caricchio R. Differential regulation of cell death programs in males and females by Poly (ADP-Ribose) Polymerase-1 and 17beta estradiol. Cell Death Dis. 2013; 4:e758.
57. Mabley JG, Horvath EM, Murthy KG, Zsengeller Z, Vaslin A, Benko R, Kollai M and Szabo C. Gender differences in the endotoxin-induced inflammatory and vascular responses: potential role of poly(ADP-ribose) polymerase activation. J Pharmacol Exp Ther. 2005; 315:812-820.
58. Baggio G, Corsini A, Floreani A, Giannini S and Zagonel V. (2013). Gender medicine: a task for the third millennium. Clinical Chemistry and Laboratory Medicine, pp. 713.
59. Miller VM and Bahn RS. Mentoring interdisciplinary research teams for the study of sex and gender differences in health and disease. Glob Adv Health Med. 2013; 2:109-112.
60. Yoon DY, Mansukhani NA, Stubbs VC, Helenowski IB, Woodruff TK and Kibbe MR. Sex bias exists in basic science and translational surgical research. Surgery. 2014; 156:508-516.
61. Beery AK and Zucker I. Sex bias in neuroscience and biomedical research. Neurosci Biobehav Rev. 2011; 35:565-572.
62. Likhite VS, Cass EI, Anderson SD, Yates JR and Nardulli AM. Interaction of estrogen receptor alpha with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. The Journal of biological chemistry. 2004; 279:16875-16882.
63. Zhang F, Wang Y, Wang L, Luo X, Huang K, Wang C, Du M, Liu F, Luo T and Huang D. Poly(ADP-ribose) polymerase 1 is a key regulator of estrogen receptor alpha-dependent gene transcription. The Journal of biological chemistry. 2013; 288:11348-11357.
64. Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M and Wagner EF. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes & Development. 1995; 9:509-520.
65. Engelward BP, Weeda G, Wyatt MD, Broekhof JL, de Wit J, Donker I, Allan JM, Gold B, Hoeijmakers JH and Samson LD. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc Natl Acad Sci U S A. 1997; 94:13087-13092.
66. Deichmann WB, LeBlanc, T.J. Determination of the approximate lethal dose with about six animals. Journal of Industrial Hygiene and Toxicology. 1943; 25:415-417.