
INTRODUCTION
Cytokine receptor-like factor 2 (CRLF2), also known as thymic stromal lymphopoietin (TSLP) receptor, is a type I cytokine receptor. The CRLF2 subunit generates the functional receptor for TSLP by forming a heterodimeric complex with interleukin-7 receptor alpha (IL7Rα) [1]. Both the cytokine and its receptor have been implicated in tumorigenesis [2]. Overexpression of CRLF2 in leukemia has been reported [3–6] and was correlated with poor outcome in pediatric acute lymphoblastic leukemia (ALL) [2, 7, 8]. However, the correlation of CRLF2 expression with the prognosis of adult ALL patients has not been fully determined.
The mechanisms of CRLF2 overexpression in leukemia are not fully understood [2]. Several studies have described the correlation between elevated CRLF2 mRNA expression and genomic lesions affecting CRLF2 [8]. CRLF2 rearrangements (IGH–CRLF2; PAR1 deletion and P2RY8–CRLF2) have been reported to result in elevated expression of wild-type CRLF2 [3–5, 7]. Overexpression of CRLF2 occurs in 15% adult and pediatric ALL [9, 10]. However, the CRLF2 rearrangement subtype is reported to account for only about half of ALL with high CRLF2 expression [11, 12]. These findings suggest that factors other than CRLF2 rearrangement can be responsible for CRLF2 overexpression. IKZF1 deletion occurs in 43% of pediatric ALL with CRLF2 overexpression [11], although it is present in ~80% of Ph-like ALL with CRLF2 rearrangement [13]. IKZF1 deletions and CRLF2 overexpression were also identified in 12% and 11%, respectively, of Japanese Ph-like ALL patients, [7]. However, it is unknown whether CRLF2 is a direct target of IKZF1 or if IKZF1 deletion is associated with increased CRLF2 expression in ALL, particularly in cases without CRLF2 rearrangement.
Recently, we reported the IKZF1 global binding profiling in ALL cells and found that IKZF1 regulates the expression of its targets through chromatin remodeling in ALL [14–17]. Our ChIP-seq data showed IKZF1 binding peaks in the promoter region of CRLF2. We also found that CK2 inhibitors could increase the tumor suppressor activity of IKZF1 and act as a functional activator of the IKZF1 protein [14–16]. However, it is still unclear whether and how IKZF1 regulates CRLF2 expression.
Here we examined CRLF2 expression in adult ALL patients without CRLF2 rearrangement, and analyzed the correlation of CRLF2 expression with prognosis in adult ALL patients. We observed high expression of CRLF2 in high-risk leukemia and this correlates with the poor clinical outcome. We also identified that CRLF2 as a direct target of IKZF1. IKZF1 binds to and suppresses CRLF2 expression in ALL leukemic cells. The CK2 inhibitor, TBB, which restores IKZF1 tumor suppressor activity, increased the suppression of CRLF2 expression and IKZF1 knockdown blocks the TBB-induced changes. Deletion of IKZF1 is significantly associated with CRLF2 overexpression in adult ALL. We also found that IKZF1 suppresses CRLF2 expression by chromatin remodeling through recruitment of H3K9me3 to the promoter. Our findings indicate that IKZF1 deletion may be responsible for the overexpression of CRLF2 in high-risk ALL without CRLF2 rearrangement. High expression of CRLF2 may work in conjunction with IKZF1 deletion to drive oncogenesis of ALL and both of them have significance in an integrated prognostic model for adult high-risk ALL.
RESULTS
High CRLF2 expression is associated with poor prognosis in adult ALL without CRLF2 rearrangement
CRLF2 is highly expressed in the reported microarray cohort study of ALL patients (Supplementary Figure 1). Here we examined CRLF2 mRNA expression in 100 newly diagnosed adult ALL patients without CRLF2 rearrangement (B-ALL 65 cases and T-ALL 35 cases). We found that the CRLF2 mRNA level is significantly higher in both B-ALL and T-ALL patients when compared to that of normal bone marrow controls (Figure 1A).
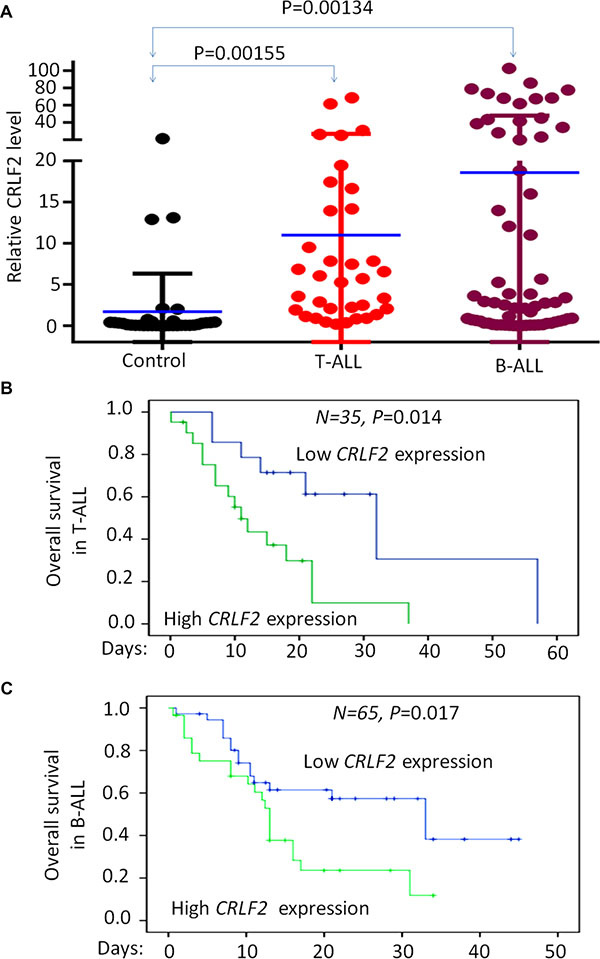
Figure 1: Correlation of CRLF2 overexpression and correlation with survival in adult ALL. (A) Comparison of CRLF2 mRNA level in B-ALL and T-ALL with normal BM control by qPCR. (B–C) CRLF2 expression with overall survival in adult T-ALL (B) and B-ALL (C).
We divided the patients into high CRLF2 expression (Quartile 1-Quartile 2) or low expression (Quartile 3-Quartile 4) groups and analyzed the relationship between CRLF2 expression and clinical characteristics in the cohort of B-ALL and T-ALL respectively (Supplementary Table S1 and S2). High CRLF2 expression in B-ALL patients showed a significant association with higher median WBC (45.8 × 109/L vs. 19.4 × 109/L, P = 0.035) and a higher percentage of WBC ≥ 30 × 109/L (85.7% vs. 38.9%, P < 0.0001), which are markers of poor prognosis in B-ALL (Supplementary Table 1). In addition, patients with high CRLF2 expression were more frequently positive for the stem cell marker CD34 (85.7% vs. 55.6%, P = 0.010) and the myeloid markers CD13 (60.7% vs. 33.3%, P = 0.029) and CD33 (59.3% vs. 30.6%, P = 0.023), which are indicative of a poor prognosis in B-ALL patients. We also observed that the high CRLF2 expression group had a higher frequency of splenomegaly (58.6% vs. 27.8%, P = 0.012), indicating extramedullary infiltration. Importantly, detection of Ikaros isoform6 (IK6), produced by the most common type of IKZF1 deletion, was statistically higher in the B-ALL patients with high CRLF2 expression (31.0% vs. 8.3%, P = 0.019)(Supplementary Table 1).
The T-ALL patients with high CRLF2 expression exhibited a trend toward higher median WBC (82.0 × 109/L vs. 24.8 × 109/L, P = 0.256) than those with low CRLF2 expression. Patients with high CRLF2 expression had lower hemoglobin (114.0 g/L vs. 144.0 g/L, P = 0.039), as well as higher percentages of blasts in peripheral blood (82.0 vs. 33.0, P = 0.032), higher rates of positivity for CD34 (71.4% vs. 21.4%, P = 0.006), and CD33 (61.9% vs. 21.4%, P = 0.036) and higher frequencies of lymph node infiltration (90.5% vs. 57.1%, P = 0.039) as compared to patients with low CRLF2 expression (Supplementary Table 2). No significant differences in CRLF2 expression were observed with respect to age, gender, platelet, bone marrow blasts or complex karyotype (Supplementary Tables 1 and 2). These data indicate that a high level of CRLF2 expression is associated with poor prognostic markers in this cohort of the adult ALL patients without CRLF2 rearrangements.
We further analyzed the overall survival (OS) in the cohort of B-ALL and T-ALL respectively. We found that cases with high CRLF2 expression showed significantly shorter OS than cases with low CRLF2 expression in both B-ALL (13.0 months vs. 33.0 months, P = 0.017) and T-ALL (11 months vs. 32.0 months, P = 0.014) (Figure 1B and 1C). These data further indicate that high CRLF2 expression is associated with poor outcome in ALL patients without CRLF2 rearrangements.
IKZF1 binds to the promoter of CRLF2 and regulates its expression in ALL
To further address the potential link between high CRLF2 expression with high-risk markers and poor outcome in adult ALL without CRLF2 rearrangement, we analyzed transcription factor motifs in the promoter region of CRLF2. As we expected, many core Ikaros binding motifs (GGGA) were identified in the promoter region of CRLF2 (Figure 2A). Notably, our ChIP-seq data showed strong binding peaks for IKZF1 in the CRLF2 promoter region [14, 15]. qChIP, assays showed significant binding of IKZF1 at the CRLF2 promoter region in both B-ALL (Nalm6 and 697) and T-ALL (CEM and MOLT-4) cells, but very weak binding in U-937 AML cells (Figure 2B). In addition, we observed that IKZF1 suppresses the promoter activity of CRLF2 by luciferase reporter assay (Figure 2C). These data indicate a direct effect of IKZF1 on transcription of CRLF2. Moreover, we found that expression of IKZF1 suppresses CRLF2 mRNA levels in both Nalm6 (Figure 3A) and CEM cells (Figure 3B). Conversely, efficient IKZF1 knockdown increased CRLF2 expression in Nalm6 (Figure 3C) and CEM cells (Figure 3D). Furthermore, treatment of Nalm6 and CEM cells with CK2 inhibitor (TBB) could suppress CRLF2 mRNA and protein levels in a dose-dependent manner (Figure 4A and 4B). CK2 knockdown with shRNA also induced the suppression of CRLF2 expression in Nalm6 (Figure 4C) and CEM (Figure 4D) cells. Importantly, IKZF1 knockdown with shRNA could block the TBB-induced decrease of CRLF2 expression in Nalm6 (Figure 4E) and CEM (Figure 4F) cells. These data indicate that CRLF2 is the direct target of IKZF1 and that IKZF1 suppresses CRLF2 expression in ALL.
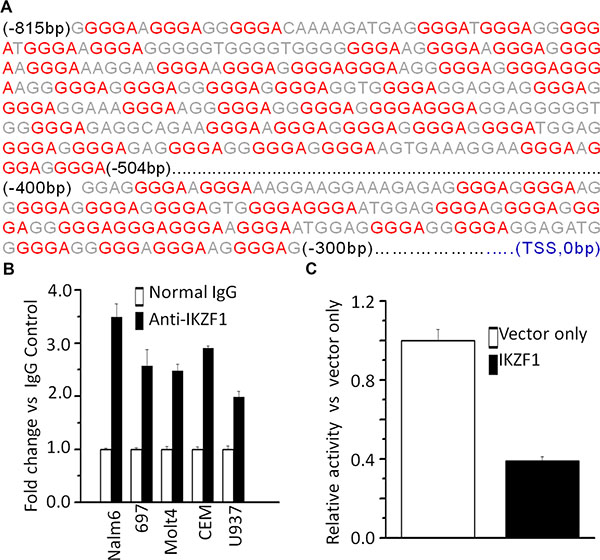
Figure 2: IKZF1 binds the promoters of CRLF2. (A) CRLF2 promoter region with conserved IKZF1 binding motif (red); (B) qChIP data for IKZF1 binding on CRLF2 promoter in leukemic cells; (C) The promoter activity of CRLF2 promoters measured by luciferase reporter assay following transfection with IKZF1 or control vector in HEK293 cells.
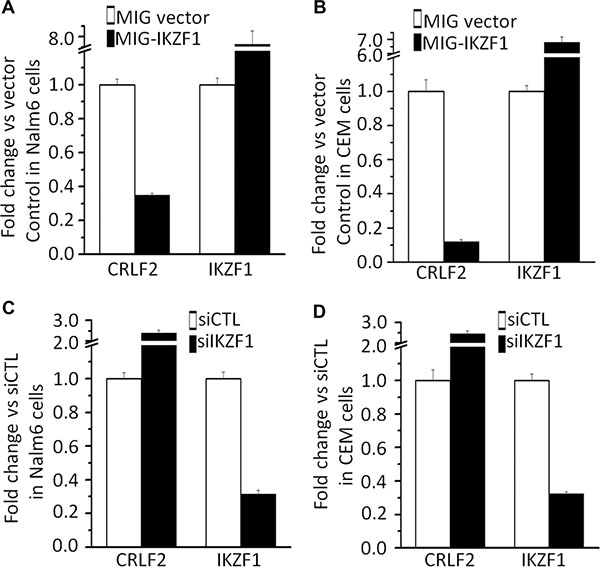
Figure 3: IKZF1 suppresses CRLF2 expression. (A–B) Expression of CRLF2 and IKZF1 in Nalm6 cells (A) and CEM T-ALL cells (B) transduced with vector containing IKZF1 as compared to control vector. (C–D) qPCR of CRLF2 and IKZF1 expression in Nalm6 cells (C) and CEM cells (D), following IKZF1 shRNA treatment as compared to scramble shRNA cells. Gene expression is determined by RT-qPCR using total RNA isolated from the cells transfected with scramble shRNA (siControl) or IKZF1 shRNA (siIKZF1) for 2 days.
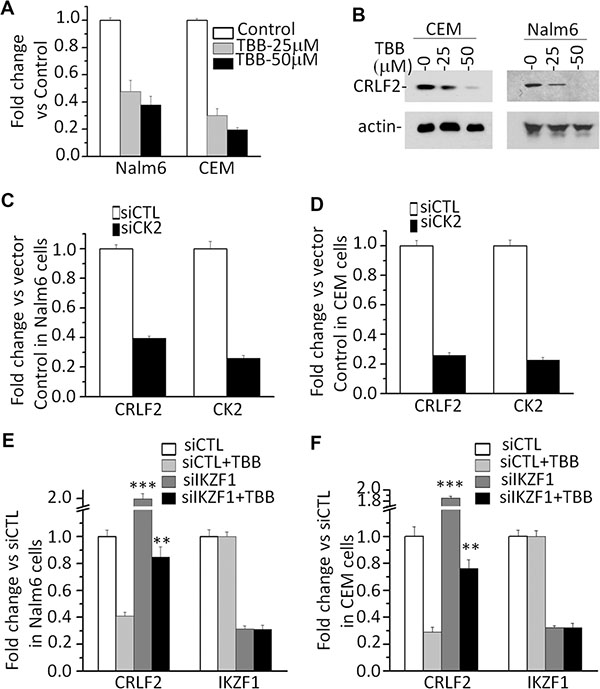
Figure 4: CK2 inhibitor-TBB suppress the expression of CRLF2 in an IKZF1-dependent manner. (A–B) Effect of CK2 inhibitor-TBB) which functions as Ikaros activator, on expression of CRLF2 mRNA level (A) and protein level (B) in Nalm6 and CEM cells with TBB treatment for 2 days. (C–D) Effect of CK2 knockdown on the expression of CRLF2 in Nalm6 (C) and CEM (D) cells. (E–F) IKZF1 knockdown rescues the TBB-induced change of CRLF2 in Nalm6 (E) and CEM (F) cells. Cells was treated with 25 μM TBB for 2 days.
Correlation of IKZF1 deletion with high high CRLF2 expression in primary ALL
To provide further supporting that that CRLF2 is a direct target of IKZF1 and that IKZF1 suppresses CRLF2 expression, we analyzed the relationship between IKZF1 deletion and high CRLF2 expression in primary ALL. We found a higher incidence of IK6, the most common protein produced by IKZF1 deletion in B-ALL patients in the high CRLF2 expression group (31% vs 8.3%, P = 0.019) (Supplementary Table 1). Moreover, we found that CRLF2 expression was significantly higher in patients with IKZF1 deletion than in patients without IKZF1 deletion (Figure 5A). These data suggest that IKZF1 deletion may contribute to high CRLF2 expression in patients. Furthermore, treatment of primary ALL cells with the CK2 inhibitor, TBB, induced an increase in IKZF1 binding at the CRLF2 promoter region in both primary B-ALL and T-ALL as compared to untreated controls (Figure 5B). Assay by qPCR also showed that TBB treatment inhibited expression of CRLF2 mRNA in a dose-dependent manner (Figure 5C). These results indicate that IKZF1 binds to the promoter of CRLF2 and that treatment with CK2 inhibitors, which can enhance IKZF1 tumor suppressor activity, resulted in suppression of CRLF2 expression in primary cells.
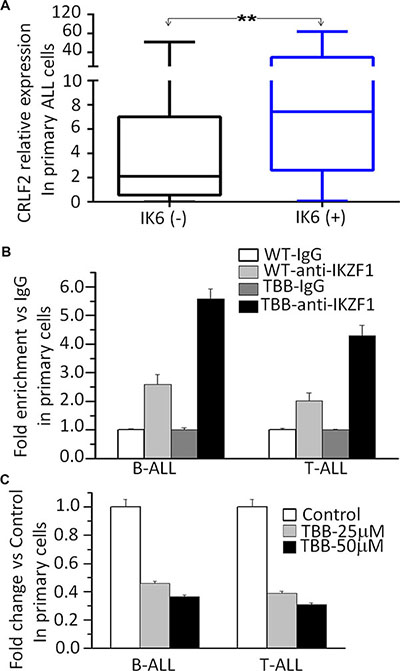
Figure 5: IKZF1 binds to the CRLF2 promoters and IKZF1 deletion results in changes of its expression in primary ALL cells. (A) Comparison of CRLF2 expression in patients with or without IKZF1 deletion, presented as CRLF2/GAPDH. The detection method for Ik6 (the most common Ikaros deletion) was done as our previously reported (23). (B) CK2 inhibitor-TBB increased the IKZF1 binding to the promoters of CRLF2 in primary B-ALL(left) and T-ALL (right) cells. (C) Effect of CK2 inhibitor-TBB functioning as IKZF1 activator on expression of CRLF2 in primary B-ALL(left) and T-ALL (right) cells with TBB treatment for 2 days.**P < 0.01.
Enhanced Ikaros activity due to CK2 inhibition increases H3K9me3 at the CRLF2 promoter
Our previous publications have reported that IKZF1 regulates gene expression through chromatin remodeling [5, 18]. To further explore the epigenetic mechanism by which IKZF1 regulates CRLF2, we performed ChIP assay and amplified the resulting CRLF2 promoter sequences with five pairs of primers as shown in Figure 6A. The results showed that TBB, which enhances IKZF1 function, significantly increases binding of IKZF1 at the CRLF2 promoter as compared with that in untreated Nalm6 B-ALL cells (WT) (Figure 6B). Moreover, TBB also clearly enhances the binding of H3K9me3 (a hallmark of heterochromatin) at the CRLF2 promoter region in Nalm6 cells (Figure 6C). Similar results were observed in the CEM T-ALL cell line (Figure 6D). However, we did not find enrichment of other histone modification markers in the CRLF2 promoter region (data not shown). Further, we found that TBB treatment increases both the binding of IKZF1 (Figure 5B) and the presence of the H3K9me3 histone modification (Figure 6E) at the CRLF2 promoter in primary ALL cells. These data indicated that enhanced IKZF1 activity suppresses CRLF2 expression via increased H3K9me3 in its promoter region.
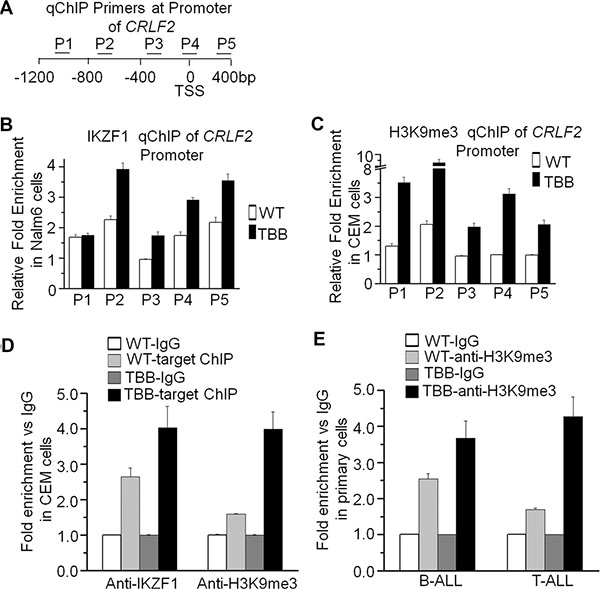
Figure 6: CK2 inhibitor-TBB by restoring Ikaros function induces transcriptionally repressive epigenetic changes at the CRLF2 promoter. (A) Schematic depiction of the qChIP primers used to analyze epigenetic changes following TBB treatment. Positions of primers used for qChIP are indicated relative to the CRLF2 transcription start site (TSS). (B–C) The qChIP analysis of the distribution of IKZF1 (B) and H3K9me3 (C) at the CRLF2 promoter in Nalm6 cells cultured with or without 50 μM TBB for 2 days. (D) The qChIP data of IKZF1 and H3K9me3 enrichment at the CRLF2 promoter in CEM T-ALL cells cultured with or without TBB. (E) The qChIP data of H3K9me3 enrichment at the CRLF2 promoter in primary ALL cells cultured with or without TBB. Graphed data are presented as mean +/– SEM of triplicates of three independent experiments.
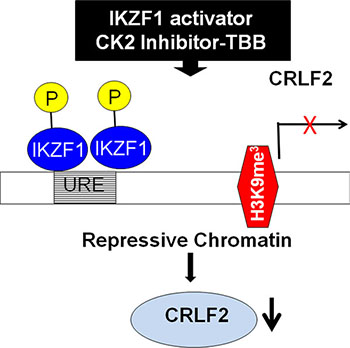
Figure 7: Proposed model of IKZF1 activator inducing suppression of CRLF2 expression.
DISCUSSION
Genetic alterations at the CRLF2 locus are associated with overexpression of CRLF2 in ALL [15–18]. CRLF2 rearrangement occurs at high frequency in Down Syndrome ALL [3, 9, 19] and in patients of Hispanic/Latino ethnicity [21]. However the rate of CRLF2 overexpression occurs at a higher frequency in patients than CRLF2 rearrangement [9, 22]. Thus CRLF2 rearrangement is unable fully to explain the mechanisms underlying CRLF2 overexpression in ALL patients. Here we provide the first report that CRLF2 is a direct target of IKZF1 and that IKZF1 regulates CRLF2 expression via induction of the H3K9me3 histone modification at its promoter (Model see Figure 7). We also show that Ikaros deletion is associated with high CRLF2 which suggests another mechanism, in addition to genetic alteration of the CRLF2 locus, is responsible for CRLF2 overexpression in ALL.
In this cohort study, we focus on ALL patients without CRLF2 rearrangement. We found that high expression of CRLF2 is correlated with unfavorable prognostic factors in the patients. Strikingly, the overall survival was significantly shorter in both T-ALL and B-ALL patients with elevated CRLF2. These observations suggested that CRLF2 overexpression is also correlated with high-risk adult ALL even when the CRLF2 locus has not undergone rearrangement.
It is worth noting that we observed expression of Ik6 in 18.5% of B-ALL without CRLF2 rearrangement and particularly that 75.0% of these IK6+ patients also showed overexpressed CRLF2. These data indicate that IKZF1 deletion with CRLF2 over expression but no CRLF2 rearrangement may distinguish a subgroup of adult ALL patients with unfavorable prognosis. This data also revealed the prognostic correlations for IKZF1 deletion and high CRLF2 expression in ALL patients. More importantly, we observed that CRLF2 expression is significantly higher in patients with Ik6, suggesting a role for the IKZF1 deletion in the elevated CRLF2 expression.
Our previous publications have demonstrated that IKZF1 exerts anti-tumor effects by regulating the expression of its target genes such as c-Myc, PI3KCD, KDM5B, etc. [14–17]. Here we observed that IKZF1 binds to the promoter region of CRLF2 and directly suppresses the promoter activity of CRLF2. Our data provide evidence that IKZF1 suppresses CRLF2 expression and conversely, that IKZF1 knockdown increases CRLF2 expression in ALL cells. We also showed that treatment with the CK2 inhibitor, TBB, which can restore/enhance Ikaros function, could increase IKZF1 binding at the CRLF2 promoter and suppress CRLF2 expression in both ALL cell lines and primary cells in an IKZF1 dependent manner. These data indicate that CRLF2 is a direct target of IKZF1 and that increasing IKZF1 activity increases the suppression of CRLF2 expression in ALL.
We previously reported that IKZF1 regulates gene expression through chromatin remodeling [15, 19]. Here, we observed that increasing IKZF1 activity via the CK2 inhibitor, TBB, increases H3K9me3, a suppressive chromatin marker, at the CRLF2 promoter. This indicates that suppression of CRLF2 expression via chromatin remodeling is also a mechanism underlying IKZF1 anti-oncogenesis in ALL, particularly high-risk ALL.
In summary, we report that CRLF2 is highly expressed and significantly correlated with poor clinical outcome in a cohort of adult ALL patients without CRLF2 rearrangements. We found that Ikaros deletion (characterized by Ik6 expression) is significantly correlated with high CRLF2 expression. We are the first to show that CRLF2 is a direct target of IKZF1, that IKZF1 regulates CRLF2 expression, and that restoration of IKZF1 activity using the CK2 inhibitor, TBB, can suppress CRLF2 expression via chromatin remodeling in ALL. Our results strongly implicate CRLF2 overexpression in conjunction with IKZF1 deletion in the oncogenesis of ALL and suggest that these should be integrated in future prognostic models for adult ALL patients.
MATERIALS AND METHODS
Patients and samples
Bone marrow (BM) samples from 100 patients with newly diagnosed ALL and without CRLF2 rearrangements (65 B-ALL and 35 T-ALL) were collected between June 2008 and June 2015 at the First Affiliated Hospital of Nanjing Medical University. Thirty normal bone marrow samples were obtained from the volunteer. Written informed consent was provided before enrollment in the study by all patients and volunteers in accordance with the Declaration of Helsinki. The study was approved by the Ethics Committee of The First Affiliated Hospital of Nanjing Medical University, Jiangsu Province Hospital, Nanjing, China. The diagnosis of ALL were made according to the morphologic, immunophenotypic, cytogenetic and molecular criteria of WHO Diagnosis and Classification of ALL (2008).
Cytogenetic and molecular analyses
Cytogenetic analysis, determination of Ik6 [23], CRLF2 rearrangements [3, 10, 21], BCR-ABL fusion gene/Ph chromosome [8, 23] were performed as previously described. qPCR was performed with qSTAR SYBR Master Mix (OriGene) on StepOne Plus Real-time PCR system (Applied Bioscience). In order to quantitate gene expression value in patients’ samples, template standards and primers against Homo sapiens gene CRLF2 (OriGene, USA) and Homo sapiens housekeeping gene GAPDH (OriGene, USA) were obtained from OriGeneTechnologies (Rockville, MD, USA). Gene expression values of the genes of interest (GOI) were achieved in each patient by a formula obtained with a scatter graph of the Ct values from the serial dilutions of template standard following manufacturer’s instruction and as previously reported [16, 24]. The expression level of the GOI was subsequently normalized to the housekeeping gene, expressed as gene expression value of GOI/GAPDH. Each experiment was performed in triplicate. All patients were placed in high or low CRLF2 expression groups (Quartile 1-Quartile 2 vs. Quartile 3-Quartile 4) and the cut-off values were determined by SPSS 17.0 [16].
The qPCR for CRLF2 expression was similarly performed as above in Nalm6 and CEM cells. The results were normalized to those obtained with 18sRNA and presented as fold induction over vector controls. Primers: 18s RNA, Sense:5′-GTAACCCGTTGAACCCCATT-3′, Antisense: 5′- CCATCCAATCGGTAGTAGCG-3′;CRLF2 Sense: 5′- GCCAGACCCGAAATCCATCT -3′, Anti-sense: 5′- CCTGGAAGTTCCCTTGGTGTAT -3′.
Cell culture reagents, plasmid construction, and retroviral gene transfer
The Nalm6 [25] and 697 [11] have been previously described. CCRF-CEM (CEM), MOLT-4 and U-937 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cell lines were cultured in RPMI 1640 medium (Cellgro, USA) supplemented with 10% fetal bovine serum (Hyclone, USA). HEK 293T cells were cultured in DMEM (Cellgro) supplemented with 10% fetal calf serum and 1% L-glutamine (Cellgro). Cells were incubated at 37°C in a humidified atmosphere of 5% CO2. Primary human B-ALL and T-ALL cells were cultured in RPMI 1640 medium (Cellgro) supplemented with 10% fetal bovine serum (Hyclone). 4, 5, 6, 7-Tetrabromobenzotriazole (TBB) was purchased from Sigma (St. Louis). Cells were cultured with or without TBB and collected for total RNA isolation. Human HA-tagged IKZF1 retroviral construct and retroviral production was described previously [14, 16, 26, 27].
Luciferase assay
The promoter of CRLF2 (–1000 bp to +200 bp) was cloned into pGL4.15 vector (Promega, WI, USA). The transient luciferase assay was performed in HEK293T cells using the Promega luciferase assay reagents and measured with luminometer following the manufacture’s instruction [14, 16]. The firefly luciferase activities were calculated as fold change relative to values obtained from pGL4.15 vector only control cells, and expressed as a percentage of pcDNA3. 1-IKZF1 transfection-induced luciferase activity versus that of pcDNA3.1 vector. All transfection and reporter assays were performed independently, in triplicate, at least three times.
Quantitative chromatin immunoprecipitation (qChIP)
The qChIP assays were performed by incubation of the chromatin with antibodies against IKZF1 [14–16], H3K9me3 (Abcam, USA) or normal rabbit IgG (Abcam) as a control [14–16, 23]. Enrichment of the ChIP sample over input was evaluated by qPCR with three or more replicates, using specific primers in the promoter region of CRLF2 as shown in Supplementary Table 3 (forward: 5′-AGGGGA AGGGAGGGGAAGGGAAAG-3′, reverse: 5′-CCCTTTCCTCCCCTCCCCTCCT-3′). Relative concen- tration of the qPCR product was presented as the fold change of the level of DNA-IKZF1 and DNA-H3K9me3 samples in comparison to controls.
IKZF1 shRNA knockdown
The Nalm6 cells were transiently transfected with human IKZF1 shRNA constructs in the GFP vector (pGFP-v-RS) (Origene) using the Neon Transfection System (Invitrogen, USA). We used ascrambled 29-mer shRNA cassette in the pGFP-v-RS vector as a control. Knockdown of IKZF1 was confirmed by measurement of IKZF1 mRNA level with qPCR [14, 16, 27]. Primers: IKZF1-F: 5′-GGCGCGGTGCTCCTCCT-3′, IKZF1-R: 5′-TCCGACACGCCCTACGACA-3′.
Western blot
Cell lysate from cells was prepared as previous reported [28]; Twenty microgram protein was boiled in SDS sample buffer for 10 minutes; the resulting samples were run on SDS-PAGE and transferred to the membrane. Membranes were blocked with 5% nonfat dry milk at room temperature for 1 h and then incubated overnight at 4°C with primary antibody (anti-CRLF2, 1:500; Santa Cruz). After being washed, the membrane was incubated with goat anti-rabbit IgG conjugated to horseradish peroxidase (1:3000) at room temperature for 2 h. The blots were developed by the enhanced chemiluminescence technique (ECL Plus, Amersham, Arlington Heights, IL) according to the manufacturer’s instructions.
Statistical analysis
We divided patients into high or low CRLF2 expression groups (Q1-Q2 vs. Q3-Q4) [16, 24]. Overall median differences between the two groups were evaluated using a Mann–Whitney U-test. Overall frequency differences in the two groups were analyzed using a χ2 test. Survival analysis was calculated using the Kaplan–Meier method. All statistical analyses were performed using the SPSS version 17.0, and P < 0.05 was considered statistically significant.
The experimental data were shown as the mean value with bars representing the standard error of the mean (SEM). Determinations of statistical significance were performed using a Student t-test for comparisons of two groups or using analysis of variance (ANOVA) for comparing multiple groups. P < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This work is supported in part by The National Natural Science Foundation of China (81270613, 30973376); Jiangsu Province Key Medical Talents (RC2011077); The Scientific Research Foundation for the Returned Overseas Chinese Scholars; State Education Ministry (39th); China Postdoctoral Science Foundation (20090461134 ); Special grade of the financial support from China Postdoctoral Science Foundation (201003598); The Six Great Talent Peak Plan of Jiangsu (2010-WS-024); The Scientific Research Foundation for the Returned Overseas Chinese Scholars; Nanjing Municipal Bureau of Personnel (2009); Southeast University Basic Research Fund (2242016k40143) (ZG). This work has been supported by Bear Necessities Pediatric Cancer Foundation; The Four Diamond Foundation of Pennsylvania State University, College of Medicine; The John Wawrynovic Leukemia Research Scholar Endowment (SD); A Hyundai Hope on Wheels award (SD and KJP); NIH awards R01CA209829 (KJP and SD), P20MD006988 and R25GM060507 (KJP).
CONFLICTS OF INTEREST
All the authors declare no conflicts of interest.
REFERENCES
1. Singh KP, Kannan K, Goel SK, Pandya KP, Shanker R. 2,5-Hexane diol induced thymic atrophy and lymphocytotoxicity in rats. Ind Health. 1983; 21:235–42.
2. Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, Borowitz MJ, Camitta BM, Carroll AJ, Devidas M, Pullen DJ, Payne-Turner D, Tasian SK, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. [Evaluation StudiesResearch Support, N.I.H., ExtramuralResearch Support, Non-U.S. Gov’t]. 2012; 119:3512–22.
3. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC,Willman CL, Mikhail FM, Meyer J, Carroll AJ, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. [Research Support, N.I.H., ExtramuralResearch Support, Non-U.S. Gov’t]. 2009; 41:1243–6.
4. Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, Shiloh R, Iacobucci I, Shochat C, Zeligson S, Cario G, Stanulla M, Strehl S, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. [Research Support, Non-U.S. Gov’t]. 2010; 115:1006–17.
5. Collins-Underwood JR, Mullighan CG. Genomic profiling of high-risk acute lymphoblastic leukemia. Leukemia. [Research Support, N.I.H., ExtramuralResearch Support, Non-U.S. Gov’tReview]. 2010; 24:1676–85.
6. Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, Chandrasekaran T, Chapiro E, Gesk S, Griffiths M, Guttery DS, Haferlach C, Harder L, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009; 114:2688–98.
7. Yamashita Y, Shimada A, Yamada T, Yamaji K, Hori T, Tsurusawa M, Watanabe A, Kikuta A, Asami K, Saito AM, Horibe K. IKZF1 and CRLF2 gene alterations correlate with poor prognosis in Japanese BCR-ABL1-negative high-risk B-cell precursor acute lymphoblastic leukemia. Pediatr Blood Cancer. [Clinical TrialMulticenter StudyResearch Support, Non-U.S. Gov’t]. 2013; 60:1587–92.
8. Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015; 125:3977–87.
9. Tasian SK, Loh ML. Understanding the biology of CRLF2-overexpressing acute lymphoblastic leukemia. Crit Rev Oncog. 2011; 16:13–24.
10. Yoda A, Yoda Y, Chiaretti S, Bar-Natan M, Mani K, RodigSJ, West N, Xiao Y, Brown JR, Mitsiades C, SattlerM, Kutok JL, DeAngelo DJ, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010; 107:252–7.
11. Findley HW, Jr., Cooper MD, Kim TH, Alvarado C, Ragab AH. Two new acute lymphoblastic leukemia cell lines with early B-cell phenotypes. Blood. 1982; 60:1305–9.
12. Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015; 12: 344–57.
13. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, Ma J, Becksfort J, Rusch M, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. [Research Support, N.I.H., ExtramuralResearch Support, Non-U.S. Gov’t]. 2014; 371:1005–15.
14. Song C, Gowda C, Pan X, Ding Y, Tong Y, Tan BH, Wang H, Muthusami S, Ge Z, Sachdev M, Amin SG, Desai D, Gowda K, Gowda R, et al. Targeting casein kinase II restores IKZF1 tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood. 2015; 126:1813–22.
15. Song C, Pan X, Ge Z, Gowda C, Ding Y, Li H, Li Z, Yochum G, Muschen M, Li Q, Payne KJ, Dovat S. Epigenetic regulation of gene expression by IKZF1, HDAC1 and Casein Kinase II in leukemia. Leukemia. 2015.
16. Ge Z, Guo X, Li J, Hartman M, Kawasawa YI, Dovat S, Song C. Clinical significance of high c-MYC and low MYCBP2 expression and their association with IKZF1 dysfunction in adult acute lymphoblastic leukemia. Oncotarget. 2015; 6:42300–11. doi: 10.18632/oncotarget.5982.
17. Song C, Li Z, Erbe AK, Savic A, Dovat S. Regulation of IKZF1 function by casein kinase 2 and protein phosphatase 1. World J Biol Chem. 2011; 2:126–31.
18. Wang H, Song C, Ding Y, Pan X, Ge Z, Tan BH, Gowda C, Sachdev M, Muthusami S, Ouyang H, Lai L, Francis OL, Morris CL, et al. Transcriptional Regulation of JARID1B/KDM5B Histone Demethylase by Ikaros,Histone Deacteylase 1 (HDAC1), and Casein Kinase 2 (CK2) in B Cell AcuteLymphoblastic Leukemia.LID-jbc.M115.679332 [pii]. J Biol Chem. 2015.
19. Malinge S, Ben-Abdelali R, Settegrana C, Radford-Weiss I, Debre M, Beldjord K, Macintyre EA, Villeval JL, Vainchenker W, Berger R, Bernard OA, Delabesse E, Penard-Lacronique V, et al. Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood. 2007; 109:2202–4..
20. Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, Chen IM, Atlas SR, Kang H, Ar K, Wilson CS, Wharton W, Murphy M, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010; 116:4874–84.
21. Harvey RC, Mullighan CG, Chen IM, Wharton W, Mikhail FM, Carroll AJ, Kang H, Liu W, Dobbin KK, Smith MA,Carroll WL, Devidas M, Bowman WP, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. [Clinical TrialResearch Support, N.I.H., ExtramuralResearch Support, Non-U.S. Gov’t]. 2010; 115:5312–21.
22. Yano M, Imamura T, Asai D, Moriya-Saito A, Suenobu S, Hasegawa D, Deguchi T, Hashii Y, Kawasaki H, Hori H, Kosaka Y, Kato K, Horibe K, et al. An overall characterization of pediatric acute lymphoblastic leukemia with CRLF2 overexpression. Genes Chromosomes Cancer. 2014; 53:815–23.
23. Liu P, Lin Z, Qian S, Qiao C, Qiu H, Wu Y, Li J, Ge Z. Expression of dominant-negative IKZF1 isoforms and associated genetic alterations in Chinese adult patients with leukemia. Ann Hematol. [Research Support, Non-U.S. Gov’t]. 2012; 91:1039–49.
24. Guo X, Zhang R, Liu J, Li M, Song C, Dovat S, Li J, Ge Z. Characterization of LEF1 High Expression and Novel Mutations in Adult Acute Lymphoblastic Leukemia. PloS one. [Research Support, Non-U.S. Gov’t]. 2015; 10:e0125429.
25. Campana D, Janossy G, Bofill M, Trejdosiewicz LK, Ma D, Hoffbrand AV, Mason DY, Lebacq AM, Forster HK. Human B cell development. I. Phenotypic differences of B lymphocytes in the bone marrow and peripheral lymphoid tissue. Journal of immunology. 1985; 134:1524–30. PubMed PMID: 3918103.
26. Popescu M, Gurel Z, Ronni T, Song C, Hung KY, Payne KJ, Dovat S. IKZF1 stability and pericentromeric localization are regulated by protein phosphatase 1. J Biol Chem. [Research Support, N.I.H., ExtramuralResearch Support, Non-U.S. Gov’t]. 2009; 284:13869–80.
27. Wang H, Song C, Gurel Z, Song N, Ma J, Ouyang H, Lai L, Payne KJ, Dovat S. Protein phosphatase 1 (PP1) and Casein Kinase II (CK2) regulate IKZF1-mediated repression of TdT in thymocytes and T-cell leukemia. Pediatr Blood Cancer. 2014; 61:2230–5.
28. van Bodegom D, Zhong J, Kopp N, Dutta C, Kim MS, Bird L, Weigert O, Tyner J, Pandey A, Yoda A, Weinstock DM. Differences in signaling through the B-cell leukemia oncoprotein CRLF2 inresponse to TSLP and through mutant JAK2. Blood. 2012; 120:2853–63.