
INTRODUCTION
Human malignant pleural mesothelioma (MPM) is a mesothelium-originated tumor most commonly involving the lungs, and more rarely the peritoneum and the vaginal membrane of the gonads. Its latency is long (15–40 years) and the prognosis very poor, with an overall survival time from diagnosis of less than one year. Extensive epidemiological data now demonstrate that the most important etiological factor is asbestos exposure. Rather intriguingly, there seems to be no correlation between length of exposure and risk of developing MPM [1]. There is no effective therapy for MPM, the commonly used treatment being pleuro-pneumectomy variably associated with neo-adjuvant and/or adjuvant chemotherapy, or radiotherapy and neo-adjuvant radiotherapy [2, 3]. A number of small Phase I/II trials using targeted therapy, i.e. inhibitors of vascular endothelial growth factor receptor (VEGFR), phosphatidylinositol 3-kinase/mammalian target of rapamycin (PI3K/mTOR), histone deacetylase and proteasome inhibitors are ongoing [4, 5, 6]. Gene therapy and immunotherapy are also actively investigated [7, 8]. Despite all these different therapeutic efforts, MPM is still a malignancy both difficult to diagnose and treat. Lack of effective treatments has spurred an active search for new therapeutic strategies to improve patients’ quality of life and prolong survival.
During the last few years several studies have shown that plasma membrane receptors for extracellular nucleotides, i.e. the P2 receptors (P2Rs), promote inflammation, cancer cell growth and metastatic spreading [9, 10, 11, 12, 13, 14]. Among the P2Rs, a special role in cancer is played by the P2X7 receptor (P2RX7 or P2X7R). We and others have shown that the P2X7R supports tumor cell growth both in vitro and in vivo, [13, 11], affords a proliferative advantage under limiting growth condition and glucose deprivation [15, 16, 17], facilitates extracellular matrix invasion [9], and stimulates VEGF secretion [13]. Moreover, the PI3K/GSK3β/HIF-1α pathway was recently identified as a main pathway responsible for P2X7R-mediated growth stimulation in human neuroblastoma cells [11]. Finally, we showed that administration of P2X7R blockers has a strong growth-inhibitory effect in several transplanted tumors such as melanoma, colon carcinoma and human and mouse neuroblastoma [13, 11]. Interestingly, we also observed that systemic administration of P2X7R blockers was more affective in tumor-bearing immunocompetent versus nude/nude mice [11], suggesting that the anti-tumor effect required an active immune response.
Expression of ATP receptors, notably P2X7R, is thought to have profound effects on host-tumor interaction since there is now very solid proof that the tumor microenvironment (TME) is enriched in ATP [18, 19, 20]. In the TME this nucleotide may promote tumor growth (for example via the P2X7R) and at the same time enhance host defenses, because tumor infiltrating immune cells also express P2X7R and other nucleotide/nucleoside receptors [20]. An ATP-rich TME can be a double-edge sword since P2X7R overstimulation can also be a trigger of cytotoxicity [21], but in tumors this untoward effect is apparently mitigated by an as yet poorly understood mechanism that uncouples P2X7R from downhill pro-apoptotic pathways otherwise active in healthy cells [22].
While P2X7R expression and function in several human tumors is well documented [12, 23], no data are so far available in human MPM. Therefore, in this study we aimed at investigating P2X7R expression in healthy mesothelial and malignant mesothelioma cells, and the effect of P2X7R inhibition on in vitro and in vivo tumor growth.
Our data show that the P2X7R is expressed in MPM cells, while it is completely absent in mesothelial cells from healthy subjects. Immortalization of human mesothelial cells with the pSV3Neo plasmid expressing the Simian Virus 40 (SV40) T-antigen (Tag) (SV40-Tag cells) caused P2X7R upregulation. Cellular responses activated by the P2X7R agonist benzoyl ATP (BzATP) were barely detectable in all mesothelial cells types investigated. Growth rate of mesothelial cells in vitro (with the notable exception of the SV40-Tag cells) was slow. Nevertheless, we were able to clearly inhibit growth by treatment with P2X7R agonists or antagonists. Growth inhibition was especially associated to release of the cytoplasmic marker lactate dehydrogenase (LDH) in SV40-Tag cells and MPP89 cells. MSTO-211H and IST-MES2 were inoculated subcutaneously (s.c.), while MPP89 were inoculated intraperitoneally (i.p.) in immunocompromised, nude/nude, mice. In all these three models, P2X7R targeting strongly reduced tumor growth. This is the first study to our knowledge showing that P2X7R blockade effectively inhibits in vivo MPM tumor growth.
RESULTS
P2X7R expression and function in healthy human mesothelial cells and in MPM cells
P2X7R expression was investigated in healthy human mesothelial cells (HMCs), in three MPM cell lines (MPP89, IST-MES2 and MSTO-211H) and in human mesothelial cells immortalized by SV40-Tag transfection (SV40-Tag cells). In HMCs P2X7R was virtually absent whether by conventional (Figure 1A) or qRT-PCR (Figure 1B). In MPP89, MSTO-211H and SV40-Tag cells, P2X7R transcript expression was comparable to that of THP-1 cells, a human leukemic cell line known to express this receptor. IST-MES2 expressed 3-4 fold less, and barely detectable, P2X7R transcript. Conventional PCR showed that MPP89 cells have a high P2X7R transcript content (Figure 1A). This finding was quantitatively confirmed by RT-PCR showing that this MPM cell line has a three fold higher P2X7R transcript level than the other cell lines, THP-1 included (Figure 1B). We also analyzed by qRT-PCR expression of the other P2X7R main splice variant, the truncated isoform named P2X7B (P2X7RB). As shown in Figure 1C, the P2X7RB isoform was virtually absent in all mesothelial cell lines. The P2X7R protein was present at low levels in HMC, IST-MES2, MSTO-211H and SV40-Tag cells cells, while it was about 2 fold higher in MPP89 cells. However, even in MPP89 cells the level of P2X7R protein expression was about half of that of THP-1 cells (Figure 1D–1E). These findings show that MPM cells express P2X7R to a low level, and therefore we anticipated that P2X7R-dependent responses should also be very low.
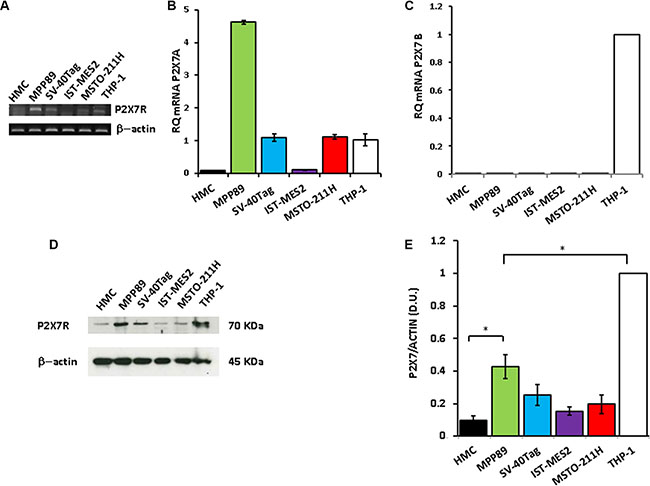
Figure 1: P2X7R expression in MPM cells. P2XR7 expression was measured by RT-PCR (A), qRT-PCR (B), and Western blotting (D–E). (C) qPCR for P2X7RB is shown. PCR, qPCR and Western blotting were performed as described in Materials and Methods. β-Actin was used as loading control. ΔΔCt was determined by assuming THP-1 cells as reference sample and human β- actin as endogenous control. For densitometry (E), P2X7R protein bands were normalized on the β- actin band. B. p < 0.001 for MPP89 versus HMC; p < 0.01 for SV-40TAG, MSTO-211H and THP-1 versus HMC. E. p < 0.001 for MPP89, SV40-Tag, MSTO-211H and IST-MES2 versus THP-1. E. *p < 0.05. Data are averages + SE from triplicate samples.
To verify this anticipation, we investigated two short term responses dependent on P2X7R stimulation: a) increase in the intracellular Ca2+ concentration, and b) permeabilization of the plasma membrane to high MW aqueous solutes (large pore opening), generally thought to be the functional signature of P2X7R. As shown in Figure 2, the semi-selective P2X7R agonist BzATP triggered a cytoplasmic Ca2+ increase in all cell lines, except HMCs, which were unresponsive throughout the whole range of BzATP concentrations tested. Ca2+ increases were rather small compared to those observed in other P2X7R-expressing cell types, but reflected the expected BzATP dose dependency, with a maximally stimulatory dose around 400 μM. The MMP89 cells were a relevant exception as BzATP concentration higher that 100 μM caused a strong inhibition of the Ca2+ influx. In all MPM cell lines the BzATP dose-dependency of the cytoplasmic Ca2+ increase was bell-shaped. This was due to progressive chelation of extracellular Ca2+ at increasing BzATP concentrations, as previously shown by us in several different cell types [24, 20]. On the basis of the level of P2X7R protein expression, we anticipated that MPP89 cells should undergo the largest Ca2+ rise, followed by the SV40-Tag cells. At variance from this anticipation, MSTO-211H cells showed a BzATP-stimulated Ca2+ rise 4-5 fold higher than that of MPP89 cells, suggesting that in these latter cells the P2X7R might be in part sequestered intracellularly and thus not fully accessible to BzATP stimulation on the plasma membrane. In the SV40-Tag cells P2X7R stimulation triggered a Ca2+ rise in the 30–40 nM range, about twice the Ca2+ increase measured in IST-MES2 cells (15–20 nM) (Figure 2C). The Ca2+ rise was almost completely obliterated by AZ10606120, except for a small and transient Ca2+ increase observed in MSTO-211H and SV-40Tag cells (Figure 2B). In all mesothelial cell types we were unable to detect any biochemical hints suggesting the opening of large conductance pore which is generally assumed to be the functional P2X7R signature, possibly because of its low expression levels in MPM. Lack of large pore opening might depend on the preferential expression of the defective P2X7RB splice variant, as described by Adinolfi et al. [25], but this was not the case as MPM expressed no P2X7RB, as shown in Figure 1C. We also analyzed the expression of other P2XRs and P2YRs, but only P2X4R, P2Y2R and P2Y11R were expressed albeit to a very low level (not shown).
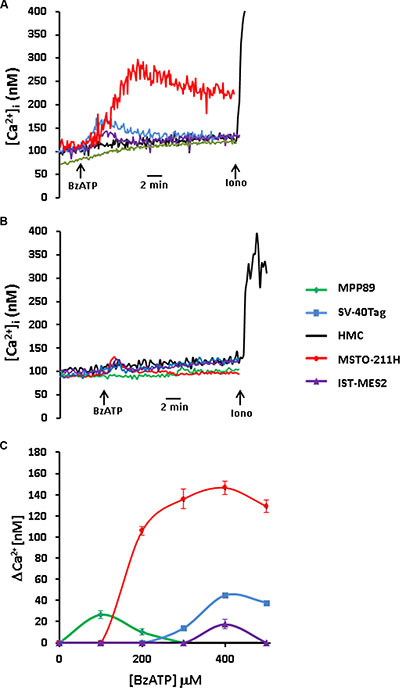
Figure 2: P2X7R-stimulated cytoplasmic Ca2+ increases in MPM cells are blocked by AZ10606120. Cytosolic Ca2+ was measured as described in Materials and Methods. (A) BzATP and ionomycin (iono) were added at the concentration of 300 and 1 μM, respectively. (B) Effect of AZ10606120 on the BzATP (300 μM)-stimulated Ca2+ increase. AZ10606120 was added to the cell suspension 5 min prior to BzATP addition. Ionomycin was 1 μM. (C) BzATP dose-response curve. Δ cytosolic Ca2+ increase (Δ [Ca2+]i) is the difference between peak and basal Ca2+ level. In the SV40-Tag curve error bars are not shown because they are smaller than the symbol. Data are averages ± SE of three to five independent determinations.
We and others have shown that basal expression or low level stimulation of the P2X7R, as opposed to sustained pharmacological stimulation, drives cell growth, and accordingly P2X7R silencing or pharmacological blockade inhibits proliferation [15, 13, 26, 27]. Growth-stimulating effect of P2X7R is enhanced in low nutrient culture conditions [16, 17]. Figure 3F shows that in the absence of serum HMCs were fully growth-arrested, while on the contrary MPM cells retained their growth ability. It is worth noting that SV40-Tag cells showed a growth kinetic that was twice as fast as that of the other MPM cell lines. As shown in panels 3A–3E, the five different mesothelial cell lines used in this study showed striking morphological differences: HMC were thin, flattened and very large (over 40 μm in diameter and 100 μm in length), with a typical mesothelial shape; SV40-Tag were fibroblast-like and about 40 μm in length. The other three cell lines were rather heterogeneous in shape and size, being IST-MES2 the smallest (with a diameter between 10 and 15 μm). We next verified whether proliferation of MPM cells was affected by P2X7R targeting.
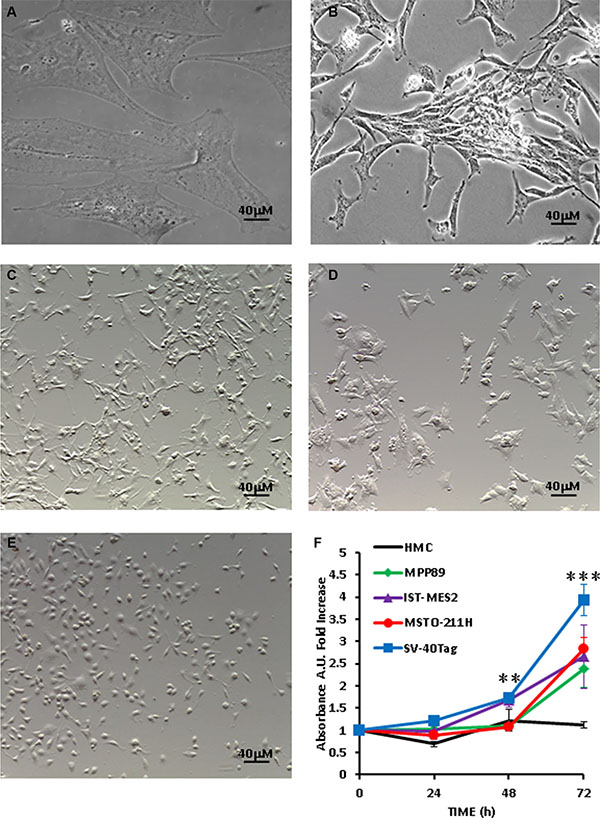
Figure 3: Growth rate and morphology of MPM cells. (A) HMCs; (B) SV40-Tag cells; (C) MPP89 cells; (D) MSTO-211H cells; (E) IST-MES2 cells. (F) Growth kinetics of HMCs and MPM cells cultured in serum-free RPMI or serum-free DMEM-F12 (MPP89) medium for 72 h; cell number was assessed by measuring crystal violet uptake at 590 nm absorbance as described in Materials and Methods. Pictures were taken with a Leica phase contrast microscope as described in Materials and Methods.
P2X7R targeting inhibits MPM cell proliferation in vitro
Responses of MPM cells to P2X7R activation or blockade were rather heterogenous. HMCs were insensitive to P2X7R targeting whether by agonists or antagonists (Figure 4A–4B). No significant decrease in cell number was observed in HMCs, nor lactic dehydrogenase (LDH) release as an index of P2X7R-dependent necrosis. At variance from HMCs, proliferation of SV40-Tag cells was sensitive to P2X7R modulation (Figure 4C–4D). The pharmacological ATP analogue BzATP strongly inhibited cell growth and caused a significant LDH release. P2XR7 blockade by the highly selective antagonist AZ10606120 or by the less specific covalent blocker oxidized ATP (oxoATP) inhibited proliferation and increased LDH release. MSTO-211H cell proliferation was fully blocked by oxoATP or by AZ10606120, but contrary to the other cell types BzATP showed growth-promoting activity (Figure 4E–4F). Of all the stimuli, only oxoATP triggered about 10% LDH release at the 72 h time point. IST-MES2 cell proliferation was blocked by P2X7R agonists or antagonists at all time points (Figure 4G–4H). In this cell type, AZ10606120 caused a severe reduction in cell number in the absence of significant LDH release, suggesting an apoptotic process. MPP89 cell proliferation was fully inhibited by BzATP or by oxoATP, while AZ10606120 had a negligible effect (Figure 4I–4J). BzATP, oxoATP and AZ10606120 had a small but significant cytotoxic effect in MPP89 cells. Despite a certain degree of heterogeneity, these results provide two relevant pieces of information: a) HMCs lack P2X7R and are fully growth arrested in the absence of serum, while MPM cells (especially SV-40-Tag) express P2X7R and retained growth ability under this condition of limited nutrient supply; b) P2X7R blockade inhibits MPM proliferation. While these data do not allow to conclude that P2X7R expression is the factor that supports growth of MPM or SV40-Tag cells in the absence of serum, the inhibitory effect of P2XR7 antagonists suggest that this receptor is involved. To gain more solid hints as to the role of P2X7R in MPM growth, we turned to the in vivo model.
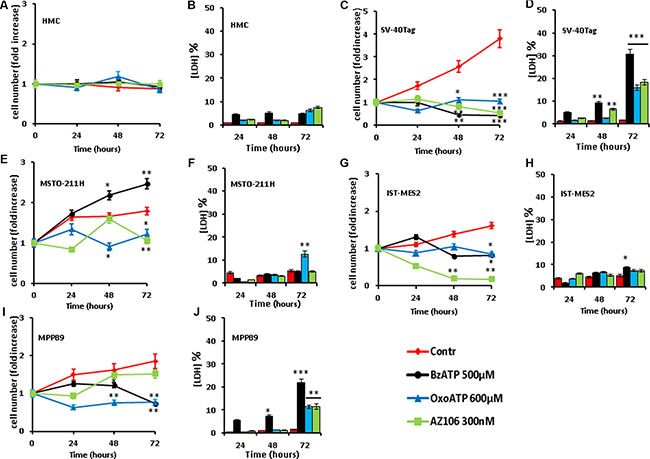
Figure 4: P2X7-targeting inhibits in vitro MPM cell growth. HMCs and MPM cells were seeded in 12-well plates as described in Materials and Methods and incubated in serum-free medium for the indicated time in the absence or presence of the various agents. Samples run in parallel were used for measurement of cell growth (panels A, C, E, G and I) and lactic dehydrogenase (LDH) release (panels B, D, F, H and J). Data are averages ± SE from nine separate determinations from three different experiments. Statistical significance of treated samples versus controls: ***p < 0.001; **p < 0.01; *p < 0.05.
P2X7R targeting inhibits in vivo mesothelioma growth
To assess the effect of P2X7R blockade on mesothelioma growth in vivo, nude/nude mice were subcutaneously (s.c.) inoculated with either MSTO-211H (Figure 5A, 5C, 5E) or IST-MES2 (Figure 5B–5D) cells and intra-mass (i.m.) injected with AZ10606120 or placebo (PBS + 0.05% DMSO). AZ-10606120 or placebo were injected every two d after tumor mass became first detectable (d six from tumor cell inoculation), and tumor mass was assessed by caliper measurement for a total of 20 (MSTO-211H) or 15 (IST-MES2) d. For the MSTO-211H inoculum, initial kinetic of tumor growth (from post-inoculum d 6 to d 10) was due both to the increase in cell number and to the tumor-associated edema (Figure 5A). After d 10, edema started to regress while the tumor mass continued its growth and bulged out of the skin surface (Figure 5E). This produced the apparent decrease in size observed in the placebo-treated mice after d 10. Treatment with AZ10606120 significantly inhibited the kinetic of tumor growth, leading to a 50% reduction of tumor size at the time of excision (Figure 5A–5C), thus confirming the strong growth inhibitory effect observed in vitro. Size of a representative tumor mass (placebo to the left) is shown in the inset in Figure 5C. IST-MES2 tumor masses were much smaller than MSTO-211H (tumor volume at d 6 post inoculum was 8–10 and 40–50 mm3 for IST-MES2 and MSTO-211H, respectively). Intra-mass administration of placebo (started at d 6) caused a growth arrest lasting up to d 12 (Figure 5B). At this time-point the placebo-treated tumor masses started to regress, shrinking to a 2–3 mm size by d 15. In the animals treated with AZ10606120 tumor regression was significantly accelerated, starting soon after drug administration. At d 15, size of excised masses from placebo- or AZ10606120-treated mice was not significantly different. IST-MES2 masses are reported as weight since small size did not allow reliable volume measurement (Figure 5D).
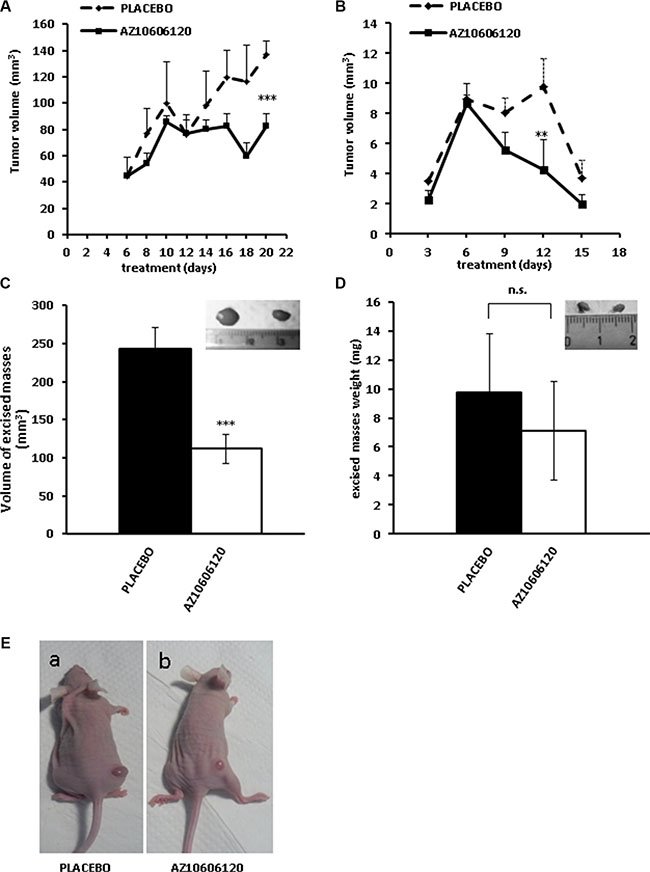
Figure 5: P2X7R-targeting inhibits growth of s.c. inoculated MPM cells. 2 × 106 MSTO-211H (panels A, C, E) or IST-MES2 (panels B, D) cells were s.c. inoculated into nude/nude mice as described in Materials and Methods. Tumors became palpable 4 d after inoculum with both MPM cell lines although the IST-MES2 mass was much smaller than the MSTO-211H mass. On post-inoculum d 6 AZ10606120 (0.7 mg/kg), or placebo (DMSO 0,05% in 100 μl PBS solution) were administered intra mass every two d for up to 20 d. Five (MSTO-211H) or 3 (IST-MES2) mice were used for AZ10606120 and placebo administration, respectively. A, B. In vivo growth kinetic. Data are averages ± SE. ***p < 0.001; **p < 0.01. B, D. Volume of excised tumors. N = 5 (MSTO-211H), or 3 (IST-MES2); ***p < 0.001; n.s., not significant. Tumor size was measured by caliper and volume calculated as described in Material and Methods. E. Pictures showing tumor size at post-inoculum d 20 in two MSTO-211H-injected mice respectively treated with placebo (a) or AZ10606120 (b). Insets in panels C and D show representative pictures of excised tumors from placebo (left)- or AZ10606120-treated (right) mice.
Tumor cell inoculation in the s.c. tissue of the hip does not faithfully reproduce the physiological environment of mesothelioma growth, therefore we investigated the effect of P2X7R targeting in MPM cells growing in the peritoneal cavity, an anatomical site where 10–20% of human mesotheliomas are commonly found [28]. To this aim we choose the MPP89 cells, which in in vitro experiments appeared to be the least sensitive to P2X7R blockade, the rationale being that should P2X7R blockade be effective also under such unfavorable in vivo conditions, this would strongly validate P2X7R targeting as a viable approach to fight mesothelioma. In order to follow tumor growth within the peritoneum, an anatomical site that is not amenable to measurement by caliper, MPP89 cells were stably transfected with a cytosolic luciferase construct (MPP89-cytLUC cells) and their growth was monitored by in vivo imaging with a total body IVIS luminometer. Mice were i.p. inoculated with the MPP89-cytLUC cells, and treated with AZ10606120 or placebo i.p. administered every two d after tumor mass became first detectable (post inoculum d 6), for a total of 12 d (Figure 6A–6B). At variance with mice injected subcutaneously, i.p.-inoculated, placebo-treated, mice showed clear signs of discomfort (irritability and aggressiveness) as early as post inoculum d 10, thus dictating an early compassionate suppression at d 12. Very interestingly, AZ10606120-treated mice showed no detectable signs of discomfort throughout the treatment. Diffusion of MPP89-cytLUC cells in the peritoneal cavity could be easily followed in both the placebo- and AZ10606120-treated groups up to post inoculum d 8. At later time points in the placebo-treated mice the peritoneal cavity became massively infiltrated with connective tissue, to an extent that luminescence emission was so perturbed to prevent an accurate analysis of tumor cell diffusion. However, despite this perturbing factor, AZ10606120-treated mice showed a more localized and fainter luminescence emission whether recorded from a ventral or a dorsal projection, suggesting a reduced diffusion of the tumor cells in the peritoneal cavity. At post-inoculum d 12, mice were sacrificed and the peritoneal cavity analyzed for luminescence emission. As shown in Figure 6C, while the placebo-treated mouse showed a diffuse infiltration of the peritoneal cavity, only a localized, low intensity, luminescence spot was detectable in the AZ10606120-treated mouse. Examination of the peritoneal cavity also revealed a diffuse connective tissue infiltrate and liver and spleen enlargement in the placebo- but not in the AZ10606120-treated mice (Figure 6D–6E).
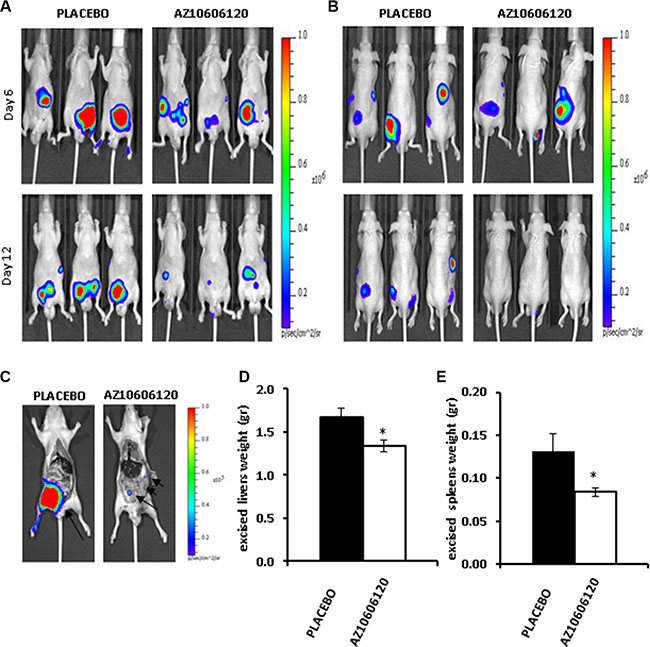
Figure 6: P2X7R-targeting inhibits growth of i.p. inoculated MPM cells. 2 × 106 MPP89/CytLUC cells were i.p. inoculated as described in Materials and Methods and on post-inoculum d 6 AZ10606120 (0.7 mg/kg), or placebo (DMSO 0,05% in 100 μl PBS solution) were administered i.p. every two d for up to 12 d. Three mice were used for AZ10606120 and placebo administration, respectively. Lumiscence emission was recorded at post-inoculum d 6 and 12 from ventral (A) and dorsal (B) projection. (C) Representative picture showing luminescence emission from the peritoneum of two mice respectively treated with placebo or AZ10606120 and sacrificed at d 12. Weight of excised livers (D) and spleen (E) from placebo and AZ10606120-treated mice. Data are averages ± SE; n = 3, *p < 0.05.
DISCUSSION
Signalling via extracellular ATP has a major role in supporting cell growth and differentiation by activating the P2YRs or P2XRs [23]. P2YRs have been traditionally credited with growth-promoting activity, as they are G- protein-coupled, and thus linked to phosphoinositide hydrolysis, Ca2+ mobilization from intracellular stores and activation of the MAPK pathway. More recent studies showed that the P2X7R also supports cell proliferation [13, 26, 29]. This was unanticipated due to the well-known cytotoxic effect due to activation of the P2X7R-associated large conductance pore [30, 31]. It is now clear that under physiological conditions, i.e. under stimulation by locally-released ATP, the P2X7R promotes a trophic response, without the untoward effects due to indiscriminate large pore opening. We have extensively characterized the mechanism responsible for the trophic P2X7R activity identifying the mitochondria as a key organelle [16, 32]. Tonic, low intensity, P2X7R activation stabilizes the mitochondrial network, and increases mitochondrial membrane potential, the Ca2+ content of the mitochondrial matrix and the overall efficiency of the oxidative phosphorylation, with the end result of increasing the total cellular ATP content. P2X7R expression also stimulates the Warburg effect, increasing glycolytic ATP generation as well as availability of carbon species for anapleurotic processes [17]. Thus, it appears that P2X7R enhances ATP generation via both the mitochondrial and glycolytic pathways, and in addition increases availability of building blocks for synthetic processes. Besides fueling energy metabolism, P2X7R also increases invasiveness and metastatic ability of cancer cells [9]. Given these P2X7R activities, it is not surprising that most, although but by no means all, malignant tumors so far tested overexpress P2X7R.
Malignant pleural mesothelioma is a peculiar tumor due to its very slow growth rate and very long latency. Therefore, on the basis of the its known growth-promoting activity we hypothesized that the P2X7R should be expressed to a low level. Our findings support this anticipation, and at the same time confirm that P2X7R, despite its low expression level, confers to MPM cells a growth advantage. In fact, HMCs kept under conditions of limited nutrient supply have a very slow growth rate over a 72 incubation time, and accordingly express little P2X7R, whether at the protein or mRNA level. However, immortalization of HMCs with the SV 40 large T antigen increases both growth rate and P2X7R expression. The three MPM cell lines investigated in the present work express P2X7R to a variable, but always very low level, and P2X7R-dependent responses are very small. Nevertheless, P2X7R inhibition by selective blockers or overstimulation by the pharmacological agonist (BzATP) inhibits growth, hinting to a growth-promoting activity of this receptor in MPM cells. While tonic P2X7R stimulation supports growth, its pharmacologic stimulation triggers cytotoxicity. Sensitivity to ATP-mediated cytotoxicity is largely dependent on the level of P2X7R expression, thus it is not surprising that MPM cell lines showed little sensitivity to P2X7R-dependent cytotoxicity in vitro. Rather, our data suggest that P2X7R pharmacological stimulation, or conversely inhibition, promote a cytostatic effect.
As it is well known, MPM is highly refractory to chemotherapy, probably due to its inherent slow growth rate. Therefore we asked if P2X7R targeting might be effective to slow down in vivo MPM growth. We tested the effect of the highly selective P2X7R drug-like antagonist AZ10606120 in three different in vivo settings: nude/nude mice inoculated s.c. with MSTO-211H or IST-MES2 cells, or i.p. with MPP89 cells. The i.p. route was investigated because we thought that it might better mimic the physiological site of MPM growth. In both cases we observed a large reduction of tumor growth in the AZ10606120-treated host. Kinetics of tumor growth in the peritoneum was of particular interest because here MPM cells diffusely infiltrated both visceral and parietal peritoneal layers causing at the same time a visible discomfort to the host. On the other hand, in the AZ10606120-treated animals there was no infiltration at all of the peritoneal layers, and the animal showed no signs of discomfort. Very few therapies are currently available to treat malignant mesothelioma, with a near 100% death rate within one year from diagnosis. Several P2X7R drug like antagonists have been extensively tested for their safety and efficacy for the treatment of chronic inflammatory diseases in over 20 PhaseII/III clinical trials [33]. Our findings might open the avenue for an exploratory trial to test the efficacy of P2X7R blockers in the therapy of malignant pleural mesothelioma.
MATERIALS AND METHODS
Cell culture and transfections
HMCs were obtained from biopsies collected from young, non-oncologic patients (age 29–35 years) affected by pneumothorax, and propagated as primary cultures in RPMI 1640 medium (EuroClone SpA, Milano, Italy) supplemented with 15% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA). MPM cell lines, MPP89, MSTO-211H and IST-MES2 from our laboratories [34], were grown in DMEM, supplemented with 2 mM L-Glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin, 10% FBS (EuroClone). Cells were maintained at 37°C in a 5% CO2-humidified atmosphere (34). The immortalized mesothelial cell line SV40-Tag was obtained by transfecting HMCs with the pSV3neo plasmid carrying the Simian Virus 40 (SV40) T-antigen (Tag) (Quiagen, Chatsworth, CA). Single-cell–derived clones were obtained by limiting dilution and kept under selection in the presence of 100 μg/ml G418-sulfate (Calbiochem, La Jolla, CA, USA) [35]. For in vivo experiments MPP89 cells were transfected with a cytosolic Luciferase (CytLUC) inserted in a pGL4.50 plasmid (Promega Italia, Milan, Italy). A Mirus kit (Mirus Bio LLC, Madison, WI, USA) was used for transfection. Single-cell-derived clones were selected in the presence of 100 μg/ml hygromycin (Sigma-Aldrich, Milano, Italy). MSTO-211H, IST-MES2 and SV40-Tag cells were cultured in RPMI medium supplemented with 15% fetal calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin. The SV40-Tag culture medium was also supplemented with 100 μg/ml G418-sulfate (Calbiochem). MPP89 and MPP89/CytLUC cells were cultured in DMEM-F12 (Sigma-Aldrich) supplemented with 15% fetal calf serum, 100 μg/ml hygromycin Sigma-Aldrich, only MPP89/CytLUC cells), 100 U/ml penicillin and 100 mg/ml streptomycin.
Cell stimulation and reagents
All experiments were performed in the absence of serum (i.e. serum-free RPMI or DMEM-F12). Cells were plated overnight in complete medium, to be replaced with serum- free medium for the next 72 hours. The P2X7R inhibitor AZ10606120 (Tocris Bioscience, Ellisville, MS, USA) was dissolved as a stock solution of 100 mM in DMSO, and used for all in vitro and in vivo experiments at a final concentration of 300 nM in sterile PBS (EuroClone).
Measurement of lactate dehydrogenase release
Cells (5 × 105) were suspended in complete medium and over-night plated in 24-well plates. Incubation medium was then replaced with serum free RPMI or DMEM-F12 supplemented with the different stimulants. At the indicated time points supernatants were collected, cleared by centrifugation (10 min at 250 g) and LDH content determined by spectrophotometry as previously described [36]. Samples lysis with 0.1% Triton ×-100 provided total LDH cell content (100% LDH release).
Cell proliferation assay and microscopy
Cell proliferation was assessed by crystal violet staining. Cells were seeded over-night in 12-well plates in complete medium at the concentration of 5 × 104 / well. Incubation medium was then replaced with serum free RPMI or DMEM-F12 at 37°C, up to 72 hours. Plates were stained with crystal violet (0.1% in distilled H2O) at each indicated time point. Crystal violet was extracted with 10% acetic acid followed by plate reading at 590 nm. Each experiment was performed in triplicate. Proliferation rate was expressed as fold increase over time zero. Pictures were taken every 24 hours with a Leica DMIL LED phase contrast microscope equipped with a 10× objective and a color camera (Leica Microsystems, Germany).
RT-PCR and qRT-PCR
Total mRNA was extracted with TRIzol Reagent and the PureLink RNA Mini Kit (Thermo Fisher Scientific), according to manufacturers’ instructions. The PCR products were analyzed on 1.2% agarose gel implemented with Gel Red dye (Thermo Fisher Scientific), and revealed with Gel Red Imaging System Documentation equipment (Alpha Innotech, San Leandro, CA, USA). qRT-PCR was performed with the High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA, USA). Samples were run in triplicate in an AB StepOne Real Time PCR (Applied Biosystems) with TaqMan Gene Expression Master Mix (Applied Biosystems) using the following TaqMan probes: NM_002562.5 (P2X7R, Life Technologies) and NM_001101.2 (human β-actin, Applied Biosystems).
Western blotting
Cells were detached by scraping and lysed in lysis buffer (300 μM sucrose, 1 mM K2HPO4, 1 mM MgSO4, 5.5 mM glucose, 20 mM HEPES (pH 7.4), 1 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride, 0.2 μg DNase, and 0.3 μg RNase, all by Sigma-Aldrich) by repeated freeze/thawing cycles. Proteins were separated on 8% Bolt-SDS precast gels (Thermo Fisher Scientific) and blotted on nitrocellulose paper (GE Healthcare Life Science). Membranes were incubated overnight with the anti-P2X7 antibody (Sigma-Aldrich), at a dilution of 1:200 in Tris-buffered saline (TBS: 50 mM Tris, 150 mM NaCl, pH 7.6), supplemented with 3% non-fat milk and 0.5% bovine serum albumin (BSA). The anti-β actin Ab was used at a dilution of 1:1000 in TBS plus 5% BSA. The secondary anti-rabbit horseradish peroxidase-conjugated Ab (Dako, Milan, Italy) was used at a dilution of 1:2000 in TBS-t buffer (TBS, 0.1% Tween-20, Sigma-Aldrich). Protein bands were revealed with an enhanced chemiluminescence detection kit (GE Healthcare Life Science).
Intracellular free Ca2+ concentration measurement
Changes in the cytoplasmic Ca2+ concentration were measured with the fluorescent indicator fura-2-acetoxymethyl ester (fura-2/AM, 4 μM) (Thermo Fisher Scientific) in a thermostat-controlled (37°C) and magnetically stirred Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies, Milan, Italy). For fura-2-AM loading cells procedure, 106/mL cells were incubated for 20 minutes in a saline solution containing 125 mM NaCl, 5 mM KCI, 1 mM MgSO4, 1 mM Na2HPO4, 5.5 mM glucose, 5 mM NaHCO3, 1 mM CaC12 and 20 mM Hepes (pH 7.4), as previously described and heretofore referred to as standard saline [37]. Measurements were carried out at the 340/380 nm excitation wavelength ratio and at an emission of 505 nm, then converted into intracellular nano-molar Ca2+ concentration by applying the following general formula:
[Ca2+]i = Kd(R – Rmin)/(Rmax – R) (Fo/Fs), where Kd is 224 nM, R is the 340:380 ratio of fluorescence of the intracellular indicator, Rmin is the 340:380 ratio of fura-2 at 1 nM Ca2+, Rmax is the 340:380 ratio of fura-2 fluorescence intensity in the presence of saturating Ca2+ concentrations , and Fo/Fs is the ratio of fura-2 fluorescence emission at 1 nM and at saturating Ca2+ concentrations at excitation 380 nm.
Pore-forming activity
P2X7-dependent pore formation and increases in plasma membrane permeability were measured by monitoring uptake of the fluorescent dye ethidium bromide (Sigma-Aldrich), as previously described (37). Briefly, cells (5 × 105/ml) were incubated in a standard saline solution without CaCl2, plus 1 mM EGTA and 20 μM ethidium bromide, in a thermostat-controlled (37°C) fluorometric cuvette under constant magnetic stirring. Uptake of the fluorescent dye was monitored over 25 min. Digitonin (100 μM) was added to achieve complete permeabilization of cells (100% fluorescence signal). Fluorescence was measured at 360 nm/580 nm excitation/emission wavelengths, with a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies).
Analysis of in vivo tumor growth
Nude/nude mice were purchased from Harlan Laboratories (Harlan Italy, S. Pietro al Natisone, Italy). 2 × 106 human mesothelioma MSTO-211H or IST-MES2 cells were (s.c.) injected into four week-old male mice. Tumor masses became detectable four d after injection. Mice were split into two groups and either treated with placebo (PBS supplemented with 0.05% DMSO) or the P2X7R inhibitor AZ10606120 (300 nM, Tocris Bioscence), administered intra-mass every two d. Tumor mass was measured every 2 d by micro-caliper. Twenty d (MSTO-211H) or 15 d (IST-MES2) after tumor inoculum mice were sacrified and masses explanted. Tumor volume was calculated with the following equation: volume = π/6 [w1 × (w2)2], where w1 = major diameter and w2 = minor diameter (13). In a second run of experiments, 2 × 106 human mesothelioma MPP89 cells stably transfected with cytoplasmic luciferase (MPP89/CytLuc cells) were (i.p.) injected into four week-old male mice. Four d after the injection, tumor masses became detectable by luminescence analysis (IVIS Lumina, Caliper-Perkin Elmer, Hopkinton, MA, USA). Thereafter, animals were split into two groups and treated with either placebo (PBS supplemented with 0.05% DMSO) or the P2X7R inhibitor AZ10606120 i.p. administered every two d. Tumor growth was monitored every 2 d by luminescence analysis. Twelve d after inoculum mice were sacrified, and livers and spleens excised and weighed. Prior to luminescence emission acquisition, mice were anesthetized (1–3% isofluorane) and i.p. injected with luciferin (150 mg/kg) (Perlin-Elmer). Luminescence emission from regions of interest was quantified by photon counting and image analysis using the Living ImageH software (Xenogen Corp, Alameda, CA). Luminescence emission was also acquired from the open peritoneal cavity of sacrified animals by directly perfusing the open abdominal cavity with D-luciferin. Animal procedures were approved by the University of Ferrara Ethic Committee and the Italian Ministry of Health in compliance with international laws and policies (European Economic Community Council Directive 86/109, OJL 358, Dec. 1, 1987, and NIH Guide for the Care and Use of Laboratory Animals).
Data analysis
All data are shown as mean ± S.E. Tests of significance were performed by unpaired t-test with Welch’s correction, using GraphPad InStat software (GraphPad, San Diego, CA, USA).
ACKNOWLEDGMENTS AND FUNDING
FDV is supported by grants from the Italian Association for Cancer Research (n. IG 13025), Telethon of Italy (n. GGP06070), ERA-NET Neuron Joint Transnational Project “Nanostroke”, the Ministry of Health of Italy (n. RF-2011-02348435), the Italian Ministry of Education, University and Research (n. RBAP11FXBC_001) and institutional funds from the University of Ferrara. FDV was also supported by a grant from the Commission of European Communities (7th Framework Program HEALTH-F2-2007-202231 “ATPBone”). MT is supported by grants from Associazione Italiana per la Ricerca sul Cancro (n. IG 16046), Fondazione Buzzi UNICEM, n. 2005, Associazione Sammarinese per la lotta contro le Leucemie e le Emopatie Maligne (A.S.L.E.M.), n. 856/15, and institutional funds from the University of Ferrara.
CONFLICTS OF INTEREST
The Authors declare no conflicts of interest.
REFERENCES
1. La Vecchia C, Boffetta P. Role of stopping exposure and recent exposure to asbestos in the risk of mesothelioma. Eur J Cancer Prev. 2012; 21:227–30.
2. Cao C, Tian D, Manganas C, Matthews P, Yan TD. Systematic review of trimodality therapy for patients with malignant pleural mesothelioma. Ann Cardiothorac Surg. 2012; 1:428–37.
3. Donahoe L, Cho J, de Perrot M. Novel induction therapies for pleural mesothelioma. Semin Thorac Cardiovasc Surg. 2014; 26:192–200.
4. Nowak AK, Millward MJ, Creaney J, Francis RJ, Dick IM, Hasani A, van der Schaaf A, Segal A, Musk AW, Byrne MJ. A phase II study of intermittent sunitinib malate as second-line therapy in progressive malignant pleural mesothelioma. J Thorac Oncol. 2012; 7:1449–56.
5. Remon J, Reguart N, Corral J, Lianes P. Malignant pleural mesothelioma: new hope in the horizon with novel therapeutic strategies. Cancer Treat Rev. 2015; 41:27–34.
6. Christoph DC, Eberhardt WE. Systemic treatment of malignant pleural mesothelioma: new agents in clinical trials raise hope of relevant improvements. Curr Opin Oncol. 2014; 26:171–81.
7. Tagawa M, Tada Y, Shimada H, Hiroshima K. Gene therapy for malignant mesothelioma: current prospects and challenges. Cancer Gene Ther. 2013; 20:150–6.
8. Izzi V, Masuelli L, Tresoldi I, Foti C, Modesti A, Bei R. Immunity and malignant mesothelioma: from mesothelial cell damage to tumor development and immune response-based therapies. Cancer Lett. 2012; 322:18–34.
9. Jelassi B, Chantome A, Alcaraz-Perez F, Baroja-Mazo A, Cayuela ML, Pelegrin P, Surprenant A, Roger S. P2X(7) receptor activation enhances SK3 channels- and cystein cathepsin-dependent cancer cells invasiveness. Oncogene 2011; 30:2108–22.
10. Roger S, Jelassi B, Couillin I, Pelegrin P, Besson P, Jiang L. Understanding the roles of the P2X7 receptor in solid tumour progression and therapeutic perspectives. Biochim Biophys Acta. 2015; 1848:2584–2602.
11. Amoroso F, Capece M, Rotondo A, Cangelosi D, Ferracin M, Franceschini A, Raffaghello L, Pistoia V, Varesio L, Adinolfi E. The P2X7 receptor is a key modulator of the PI3K/GSK3beta/VEGF signaling network: evidence in experimental neuroblastoma. Oncogene. 2015; 34:5240–5251.
12. Di Virgilio F. Purines, purinergic receptors, and cancer. Cancer Res 2012; 72:5441–7.
13. Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, Bianchi G, Kroemer G, Pistoia V, Di Virgilio F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012; 72:2957–69.
14. Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell. 2013; 24:130–7.
15. Baricordi OR, Melchiorri L, Adinolfi E, Falzoni S, Chiozzi P, Buell G, Di Virgilio F. Increased proliferation rate of lymphoid cells transfected with the P2X(7) ATP receptor. J Biol Chem. 1999; 274:33206–8.
16. Adinolfi E, Callegari MG, Ferrari D, Bolognesi C, Minelli M, Wieckowski MR, Pinton P, Rizzuto R, Di Virgilio F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol Biol Cell. 2005; 16:3260–72.
17. Amoroso F, Falzoni S, Adinolfi E, Ferrari D, Di Virgilio F. The P2X7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis. 2012; 3:e370.
18. Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS ONE. 2008; 3:e2599.
19. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011; 334:1573–7.
20. Bianchi G, Vuerich M, Pellegatti P, Marimpietri D, Emionite L, Marigo I, Bronte V, Di Virgilio F, Pistoia V, Raffaghello L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014; 5:e1135.
21. Di Virgilio F, Chiozzi P, Falzoni S, Ferrari D, Sanz JM, Venketaraman V, Baricordi OR. Cytolytic P2X purinoceptors. Cell Death Differ. 1998; 5:191–9.
22. Raffaghello L, Chiozzi P, Falzoni S, Di Virgilio F, Pistoia V. The P2X7 receptor sustains the growth of human neuroblastoma cells through a substance P-dependent mechanism. Cancer Res. 2006; 66:907–14.
23. Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinergic Signal. 2013; 9:491–540.
24. Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi-Castagnoli P, Di Virgilio F. Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol. 1996; 156:1531–9.
25. Adinolfi E, Cirillo M, Woltersdorf R, Falzoni S, Chiozzi P, Pellegatti P, Callegari MG, Sandonà D, Markwardt F, Schmalzing G, Di Virgilio F.Trophic activity of a naturally occurring truncated isoform of the P2X7 receptor. FASEB J. 2010; 24:3393–404.
26. Vazquez-Cuevas FG, Martinez-Ramirez AS, Robles-Martinez L, Garay E, Garcia-Carranca A, Perez-Montiel D, Castañeda-García C, Arellano RO. Paracrine stimulation of P2X7 receptor by ATP activates a proliferative pathway in ovarian carcinoma cells. J Cell Biochem. 2014; 115:1955–66.
27. Qiu Y, Li WH, Zhang HQ, Liu Y, Tian XX, Fang WG. P2X7 mediates ATP-driven invasiveness in prostate cancer cells. PLoS One. 2014; 9:e114371.
28. Asensio JA, Goldblatt P, Thomford NR. Primary malignant peritoneal mesothelioma. A report of seven cases and a review of the literature. Arch Surg. 1990; 125:1477–81.
29. Ledderose C, Bao Y, Ledderose S, Woehrle T, Heinisch M, Yip L, Zhang J, Robson SC, Shapiro NI, Junger WG. Mitochondrial dysfunction, depleted purinergic signaling, and defective T cell vigilance and immune defense. J Infect Dis. 2016; 213:456–464.
30. Pizzo P, Zanovello P, Bronte V, Di Virgilio F. Extracellular ATP causes lysis of mouse thymocytes and activates a plasma membrane ion channel. Biochem J 1991; 274:139–44.
31. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996; 272:735–8.
32. Di Virgilio F., Ferrari D, Adinolfi E. P2X(7): a growth-promoting receptor-implications for cancer. Purinergic Signal 2009; 5:251–6.
33. Arulkumaran N, Unwin RJ, Tam FW. A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs 2011; 20:897–915.
34. Balatti V, Maniero S, Ferracin M, Veronese A, Negrini M, Ferrocci G, Martini F, Tognon MG. MicroRNAs dysregulation in human malignant pleural mesothelioma. J Thorac Oncol 2011; 6:844–51.
35. Cacciotti P, Libener R, Betta P, Martini F, Porta C, Procopio A, Strizzi L, Penengo L, Tognon M, Mutti L, Gaudino G. SV40 replication in human mesothelial cells induces HGF/Met receptor activation: a model for viral-related carcinogenesis of human malignant mesothelioma. Proc Natl Acad Sci USA 2001; 98:12032–7.
36. Chiozzi P, Murgia M, Falzoni S, Ferrari D, Di Virgilio F. Role of the purinergic P2Z receptor in spontaneous cell death in J774 macrophage cultures. Biochem Biophys Res Commun 1996; 218:176–81.
37. Falzoni S, Munerati M, Ferrari D, Spisani S, Moretti S, Di Virgilio F. The purinergic P2Z receptor of human macrophage cells. Characterization and possible physiological role. J Clin Invest 1995; 95:1207–16.