
INTRODUCTION
Familial adenomatous polyposis (FAP) [MIM# 175100] is a familial precancerous condition characterized by the presence of numerous colorectal adenomas or polyps, with an incidence of 3-10/100,000 [1]. It is an autosomal dominant colon cancer predisposition syndrome accounting for approximately 1% of all colorectal cancers (CRC). FAP invariably results in colorectal cancer (CRC), if not detected and diagnosed early and the colon removed by total proctocolectomy [2]. Colorectal adenomas usually start to appear at the third decade of life and if untreated, gradually become symptomatic with presence of numerous colorectal adenomas or polyps by the fourth decade of life [3]. Apart from colorectal adenomas, extracolonic manifestations such as desmoids tumors, osteomas, dental abnormalities, congenital hypertrophy of the retinal pigment epithelium (CHRPE), lipomas, epidermoid cysts and upper gastrointestinal polyps may also develop in patients with FAP [4]. In addition, it has been found that FAP is also associated with thyroid cancer, brain cancer and cancers of hepatobiliary tract. On the basis of the number of colorectal adenomas or polyps and the age of onset of the patient, FAP is categorized into two types, namely; the classical FAP (CFAP) and the attenuated FAP (AFAP) [5]. Patients with CFAP usually manifest more than 100 colorectal adenomatous polyps, generally start to develop at the third decade of the life. Patients with AFAP characteristically have 10–100 colorectal adenomatous polyps, generally start to develop at the fourth decade of life. [6].
Germline mutations of the APC gene are associated with FAP. APC is a tumor suppressor gene [7, 8], associated with cell adhesion, transcriptional activation, cell migration, and apoptosis [9, 10]. The APC protein is a multi-functional molecule comprises of eight known functional subdomains involved in the regulation of cell adhesion, polarization, and migration. The main function of the APC protein is to regulate the β-catenin protein level. Mutations of APC result in the accumulation of the β-catenin protein in cytoplasm through the activation of other transcription factors including Tcf, which in turn causes aberrant activation of the canonical Wnt signaling pathway leading to uncontrolled cell proliferation, progression and development of colon cancer [11]. According to the previous published reports, the majority of pathogenic APC germline mutations belong to three categories, nonsense/frameshift mutations, splice sites mutations and deep intronic deletions. Nonsense/frameshift mutations splice sites mutations and deep intronic deletions of the APC gene lead to large genomic rearrangements, resulting in the formation of truncated APC proteins [12]. Although, it has been found that in few cases, point mutations or missense variants within the coding sequence also result in the formation of alternative transcripts due to aberrant splicing [13, 14]. It has been reported that 2% of all germline APC gene mutations are large genomic deletions [15]. Moreover, in HGMD, 1000 different APC germline mutations have been reported till date (http://www.hgmd.cf.ac.uk/ac). In addition, over 194 unique APC germline mutations from 191 patients have been reported in the Chinese population with 76 small deletions (75 small deletions are in coding region and 1 deletion in intron) (http://www.genomed.org/lovd2/home.php?select_db=APC). In the Chinese population, there was no large deletion of APC gene has previously been reported. However, the 5’end of the APC gene appears to be the most common site for germline mutations. The codon 1309 of the APC gene is a mutation cluster region (MCR) with a higher frequency of APC germline mutations. The location of the mutation in the APC gene is very significant as it is directly correlated with the phenotypic spectrum of the disease, age of onset and the appearance of extracolonic manifestations in FAP patients [16].
Here, in order to identify the molecular basis of FAP in the proband and in all the affected members of this five generation Chinese family, we screened a panel of 14 genes (APC, MLH1, MSH2, MSH6, PMS2, AXIN2, BMPR1A, EPCAM, MLH3, MUTYH, PMS1, PTEN, SMAD4, STK11) associated with colorectal cancer by targeted next-generation sequencing and Quantitative Real-Time PCR (qPCR). We identified a novel heterozygous large germline deletion of APC gene segregating with FAP phenotype among all the FAP patients in this five generation Chinese family, with autosomal dominant inheritance.
RESULTS
Family recruitment and clinical examination
We identified a five generation Chinese pedigree with 23 members, among whom five individuals were affected by FAP including two with CRC (Figure 1). Another 2 affected family members (II-1 and II-3) had died from CRC. In Table 1, we have described the detailed and comprehensive clinical information for all the affected and unaffected members in this family. A comprehensive and comparative colonoscopy and histopathology of colon and rectum for all the affected family members along with an unaffected member (III-2) are shown in Figure 2.
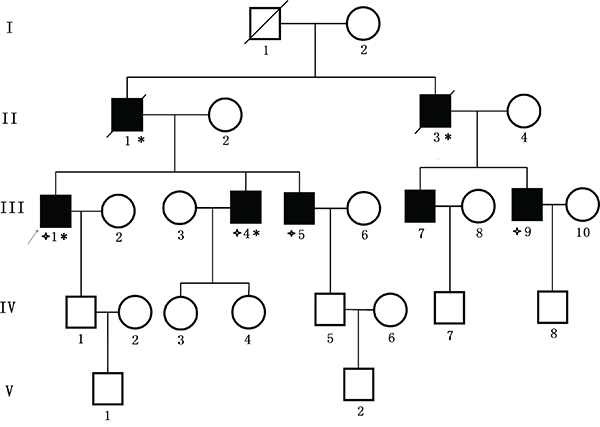
Figure 1: Pedigree structure of the Chinese family with familial adenomatous polyposis. Family members with FAP are indicated with Shading. Squares and circles denoted males and females respectively. Individuals labeled with a solidus were deceased. Family members with CRC are indicated with asterisks. Family members detected with APC gene deletion are indicated by “four angled star”. Roman numerals indicate generations. Arrow indicates the proband (III-1).
Table 1: Clinical characteristics of all the affected and unaffected family members found in our study
Family ID |
Sex |
Exon Deletion |
Present Age (Years) |
Age at Diagnosis |
No. of Polyps |
CRC Age |
Extra-colonic Features |
|---|---|---|---|---|---|---|---|
I-1 |
M |
- |
Died (73) |
- |
- |
- |
- |
I-2 |
F |
WT |
70 |
- |
- |
- |
- |
II-1 |
M |
- |
Died (69) |
- |
- |
- |
- |
II-2 |
F |
WT |
76 |
- |
- |
- |
- |
II-3 |
M |
- |
Died (56) |
- |
- |
- |
- |
II-4 |
F |
WT |
70 |
- |
- |
- |
- |
III-1 |
M |
Del |
53 |
50 |
~1000 |
53 |
- |
III-2 |
F |
WT |
52 |
- |
- |
- |
- |
III-3 |
F |
WT |
51 |
- |
- |
52 |
- |
III-4 |
M |
Del |
50 |
49 |
~1000 |
- |
- |
III-5 |
M |
Del |
48 |
46 |
~500 |
- |
- |
III-6 |
F |
WT |
48 |
- |
- |
- |
- |
III-7 |
M |
(Sample Unavailable) |
42 |
42 |
~300 |
- |
- |
III-8 |
F |
WT |
40 |
- |
- |
- |
- |
III-9 |
M |
Del |
38 |
38 |
>100 |
- |
- |
III-10 |
F |
WT |
37 |
- |
- |
- |
- |
IV-1 |
M |
- |
36 |
- |
- |
- |
- |
IV-2 |
F |
- |
35 |
- |
- |
- |
- |
IV-3 |
F |
- |
27 |
- |
- |
- |
- |
IV-4 |
F |
- |
14 |
- |
- |
- |
- |
IV-5 |
M |
- |
27 |
- |
- |
- |
- |
IV-6 |
F |
- |
26 |
- |
- |
- |
- |
IV-7 |
M |
- |
6 |
- |
- |
- |
- |
IV-8 |
M |
- |
17 |
- |
- |
- |
- |
V-1 |
M |
- |
5 |
- |
- |
- |
- |
V-2 |
M |
- |
1 |
- |
- |
- |
- |
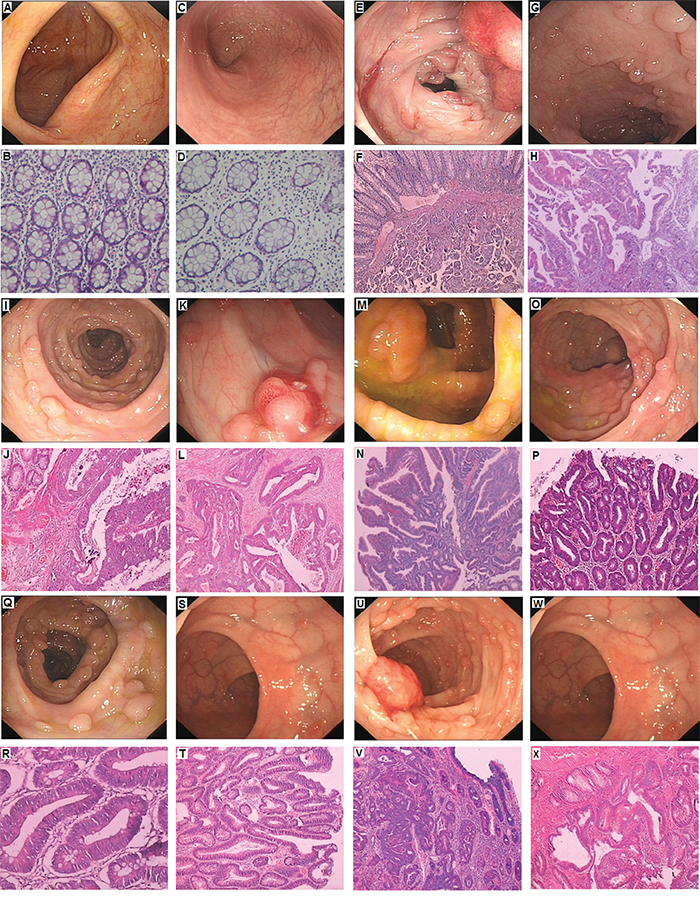
Figure 2: Clinical Description. A-B. No polyps in the colon of unaffected member with normal histology (III-2). C-D. No polyps in the rectum of unaffected member with normal histology(III-2). E-F. Polyps in the colon, Cancer invading in the submucosa in proband (III-1); G-H. Polyps of rectum showing Low grade intraepithelial neoplasia (dysplasia), the gland of local lesion showing High grade intraepithelial neoplasia (dysplasia) in proband (III-1). I-J. Polyps of colon showing High grade intraepithelial neoplasia (dysplasia) in III-4; K-L. Polyps of rectum, adenocarcinoma in III-4. M-N. Polyps of colon is Low grade intraepithelial neoplasia (dysplasia), the gland of local lesion is High grade intraepithelial neoplasia (dysplasia) in III-5; O-P. Polyps of rectum are Low grade intraepithelial neoplasia (dysplasia) in III-5; Q-R. Polyps of colon are Low grade intraepithelial neoplasia (dysplasia) in III-7; S-T. Polyps of rectum are Low grade intraepithelial neoplasia (dysplasia) in III-7; U-V. Polyps of colon are High grade intraepithelial neoplasia (dysplasia) in III-9; W-X. Polyps of rectum are Low grade intraepithelial neoplasia (dysplasia) in III-9.
Identification and characterization of candidate mutation
A heterozygous novel large deletion; [exon5-exon16; c.423_8532del]in APC gene [NCBI Reference sequence NM_000038.3] was identified in proband (III-1) by targeted next generation sequencing. This heterozygous novel large deletion co-segregated with the FAP phenotypes in the proband (III-1) and amongst all the affected family (III-4, III-5 and III-9) members, (sample of III-7 was unavailable) but absent in the unaffected family members. We did not detect this mutation in the normal control of the same ethnic origin, gender and age range.
Confirmation of the novel large deletion by quantitative real-time PCR (qPCR)
The relative DNA copy number for the APC 5, 9, 12, 16 exons was measured by quantitative real-time PCR (qPCR) in the proband (III-1) and in all the affected family members (III-4, III-5 and III-9) (Sample of III-7 was unavailable). As this is a large deletion, for validation of the result, qPCR was undertaken in the proband (III-1) and in all affected family members [III-4, III-5 and III-9], an unaffected family member (III-2) with actin being used as a control [sample of III-7 was unavailable]. For exon5, a normal negative control and unaffected family members (III-2) gave an amplification level of 1.0, while the relative amplification from the proband (III-1) and in all the affected family members [III-4, III-5 and III-9] was approximately 0.48 (Figure 3A). For exon9, a normal negative control and unaffected family members (III-2) gave an amplification level of 1.0, but the relative amplification from the proband (III-1) and in all the affected family members [III-4, III-5 and III-9] was approximately 0.5 (Figure 3B). For exon12, a normal negative control and unaffected family members (III-2) gave an amplification level of 1.0, while the relative amplification from the proband (III-1) and in all the affected family members [III-4, III-5 and III-9] was approximately 0.51 (Figure 3C). For exon16, a normal negative control and unaffected family members (III-2) gave an amplification level of 1.0, but the relative amplification from the proband (III-1) and in all the affected family members [III-4, III-5 and III-9] was approximately 0.48 (Figure 3D). There was a significant difference between affected family members [III-1 (proband), III-4, III-5 and III-9], unaffected family members (III-2) and normal control, suggesting that there was a heterozygous deletion of exon5-16 of APC in the proband (III-1) and in all the affected family members (III-4, III-5and III-9). Hence, q-PCR data suggest that proband (III-1) and affected family members (III-4, III-5 and III-9) have a novel heterozygous deletion of exon5-16 of the APC gene that is associated with FAP in this family.
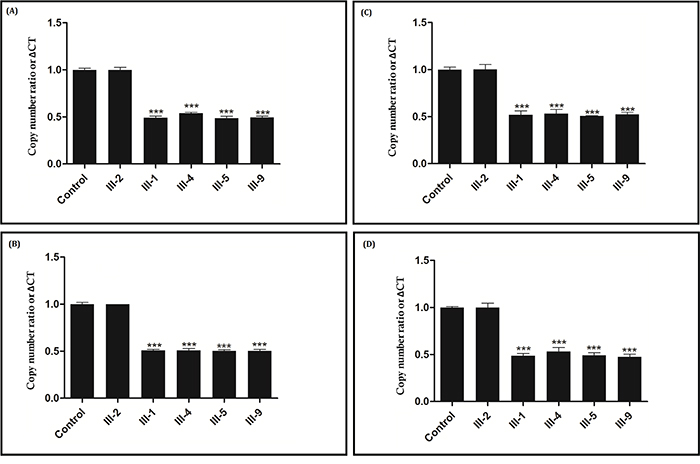
Figure 3: Verification of the novel heterozygous large deletion in III-1, III-4, III-5 and III-9 by quantitative real-time PCR (qPCR). Copy number ratio of the exon 5 Figure 3A., exon 9 Figure 3B., exon 12 Figure 3C. and exon 16 Figure 3D. in THE control and in A normal family member (III-2) is nearly 1, and the Copy number ratio in proband (III-1) and other affected family members (III-4, III-5 and III-9) is approximately half. Actin DNA was used as a loading control.
DISCUSSION
In our study, we found a heterozygous novel large deletion (exon5-exon16; c.423_8532del) [NCBI Reference sequence NM_000038.3] of the human APC gene in the proband (III-1) and among all the FAP-affected family members [III-4, III-5 and III-9] in a five generation Chinese family. This heterozygous novel large deletion of APC gene results in the formation of truncated APC protein with complete loss of the armadillo repeat domain,β-catenin binding site, microtubule binding site, EBI Domain, hDLG binding site and PDZ binding domain (Figure 4). This heterozygous novel large deletion of APC gene is not present in the ExAC database.
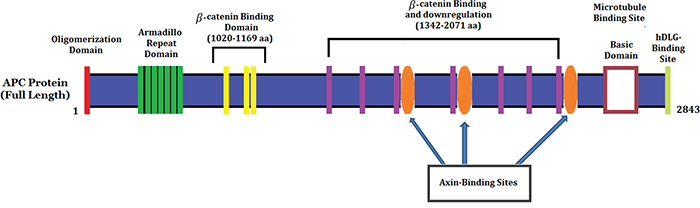
Figure 4: Schematic Diagram of the APC protein structure with functional domains.
In this Chinese family, diagnosis of FAP has been done according to a review of clinical report, endoscopic and histo-pathological data. However, the presence of specific clinical symptoms (colorectal adenomas/polyposis) and the autosomal dominant mode of inheritance allow the diagnosis of the disease in this family as FAP. In our study, the heterozygous novel large deletion of APC gene in this Chinese family manifests with colorectal adenomas (polyps) without any extra-colonic manifestations.
Large deletions of individual genes or single exon or several exons represent a well-known molecular basis of hereditary nonpolyposis colorectal cancer (HNPCC) [17, 18]. However, only 47 whole APC gene deletions have been published. In addition, most of the cases with whole APC gene deletions were diagnosed as classic FAP. Moreover, in only one case with a whole APC gene deletion found to be associated with attenuated phenotype [19]. Until now, 43 partial APC gene deletions have been reported to be associated with FAP. Among all the partial deletion of APC exons cases, deletions of exons 14 (6 probands), 14–15 (5 probands), 15 (4 probands), 8–15 (4 probands) and 9–15 (4 probands) are the most frequent. The majority of the cases with partial APC gene deletions were also associated with classic FAP. Partial deletions of APC gene are of either in-frame or out-of-frame, which correlates with the phenotype of FAP patients. APC gene in-frame deletions result in the formation of a shorter but possibly functional APC protein, so the resulting phenotype of the FAP is milder. The previously reported in-frame or out-of-frame deletions of APC gene never include the promoter region and exon 15 because the promoter region of APC gene is important for initiation of transcription and exon 15 is necessary for regulating the turnover and localization of β-catenin [20, 21].
APC gene mutation
A large number of pathogenic APC gene mutations have been reported in different countries and ethnic groups [16]. According to the HGMD dataset, small deletions account for the majority of APC gene mutations, resulting in alteration of the open reading frame followed by the formation of a truncated gene product. Until now, 275 sequence variants of the APC gene have been reported in the Chinese population. Among these, 194 are unique variants were identified out of 191 individuals. Among the 275 sequence variants of the APC gene, the majority is substitutions (164), but frameshift (101), nonsense (83), deletion (76), insertion (33) and inversion (2) are also reported (http://www.genomed.org/lovd2/variants_statistics.php).
Previous reports had not described large deletions of the APC gene in the Chinese population (http://www.genomed.org/lovd2/variants.php?select_db=APC&action= view_all&view=DNA_del).
According to a very recent report, a novel deletion in exon 14–15(c.1936-2148del) and intron 14 was described as causing FAP without any extracolonic manifestation in a Chinese family [1].
Genotype–phenotype correlation
Genotype–phenotype correlation studies are very significant and enable us to define the most likely phenotype to be associated with a given mutation. The identification and characterization of APC mutation carriers with a well diagnosed phenotype will allow us to establish specific surveillance programs and prophylactic surgical treatment. APC germline mutations, affecting the mutation cluster region (MCR), are correlated with more severe phenotype and early age of onset in FAP patients [23–27].
In conclusion, the present study describes a heterozygous novel large deletion mutation in APC gene in a five generation Chinese family with FAP. Our study expands the spectrum of the germline mutations of APC gene in the Chinese population. This novel finding contributes to a more comprehensive database of germline mutations of APC gene that could be used for the molecular diagnosis, risk assessment, susceptibility of the disease for the FAP patients.
MATERIALS AND METHODS
Ethical statement
Family members of this five generation Chinese family have given written informed consent as they are participating in this study. The Ethical Committee of the Tianjin Union Medical Center, China, reviewed and approved our study protocol in compliance with the Helsinki declaration. Diagnosis of the patients for FAP has done by oncologists, on the basis of clinical test reports and detailed family pedigree.
Patients and pedigree
A five generation Chinese family with FAP (Figure 1), diagnosed and treated in the Department of Colorectal Surgery, Tianjin Union Medical Center, 300121, China, were enrolled in our study. Clinical diagnosis of FAP was established in this family by endoscopic screening after the proband (III-1) presented to Tianjin Union Medical Center with CRC. The diagnostic standard or criteria for patients with FAP was as follows: (1) patients having >100 colorectal adenomas or polyps and (2) at least 20 synchronous colorectal adenomas or polyps in patients with a positive family history of FAP.
Targeted exome-based next-generation sequencing and variant identification
DNA samples obtained from the proband (III-1) were sequenced using target exome-based next-generation sequencing. Roche NimbleGen’s (Madison, USA) custom Sequence Capture Human Array was used to designed to capture 98480 kb of targeted sequence, covering 181 exons and flanking sequence (including the 100 bp of introns) of 14 genes (APC, MLH1, MSH2, MSH6, PMS2, AXIN2, BMPR1A, EPCAM, MLH3, MUTYH, PMS1, PTEN, SMAD4, STK11) which is associated colorectal cancer (CRC) and yielded an average of 6366534 reads per sample, with approximately 68.78% mapping to the targeted regions. The average sequencing depth of the target area is 464.68% with 99.46% coverage. The procedure for preparation of libraries was consistent with standard operating protocols published previously. In each pooling batch, 10 to 33 samples were sequenced simultaneously on Illumina HiSeq 2500 Analyzers (Illumina, San Diego, USA) for 90 cycles (specially designed by us for this study). Image analysis, error estimation, and base calling were performed using Illumina Pipeline software (version 1.3.4) to generate raw data. The raw reads were screened to generate – clean reads‖ followed by established filtering criteria. Clean reads with a length of 90 bp were aligned to the reference human genome from the NCBI database (Build 37) using the Burrows Wheeler Aligner (BWA) Multi-Vision software package with output files in - bam‖ format. The bamdata were used for reads coverage in the target region and sequencing depth computation, SNP and INDEL calling, and CNV detection. First, a novel three-step computational frame work for CNV was applied. Then, SNPs and INDELs were called using SOAPsnp software and Sam tools pileup software, respectively. A SNP or INDEL was be filtered if it could not follow the criterion: supported by at least 10 reads and >20% of the total reads. The frequency filter was set at 0.05. If a SNP frequency was more than 0.05 in any of the four databases (dbSNP, Hapmap, 1000 Genomes Project, the 124 healthy reference samples sequenced in this study), it would be regarded as a polymorphism, but not a causative mutation.
Last, SNVs were retrieved in The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and the Leiden Open Variation Database (http://www.lovd.nl/3.0/home), and then labeled as reported or novel.
Quantitative real-time PCR (qPCR)
In order to validate the result of targeted next generation sequencing and further quantify the DNA copy number change for the APC gene q-PCR was undertaken. The relative DNA copy number for the APC 5, 9, 12, 16 exons were measured by quantitative real-time PCR (qPCR) using an ABI 7900HT Real-time PCR system (Life Technologies, Carlsbad, CA, USA) and HS qPCR Master Mix, according to the manufacturer’s instructions. The primers used for amplifying APC exons were listed in Table 2.
Table 2: Detailed primer sequence for Real-time PCR
Primer |
Primer sequence |
Size/bp |
Tm/°C |
|---|---|---|---|
Exon 5_Forward |
5’-TCATTGCTTCTTGCTGATCTTGAC-3’ |
74 |
59.8 |
Exon 5_Reverse |
5’-GTGAGATTCTGAAGTTGAGCG-3’ |
74 |
57 |
Exon 9_Forward |
5’-GGTTCAACTACACGAATGGACCATGA-3’ |
95 |
60 |
Exon 9_Reverse |
5’-GTTCCCAGATGACTTGTCAGCCTTC-3’ |
95 |
60.2 |
Exon 12_Forward |
5’-GTGGACTGTGAAATGTATGGGCT-3’ |
71 |
60.8 |
Exon 12_Reverse |
5’-GCCATTCCAGCATATCGTCTTAG-3’ |
71 |
59.3 |
Exon 16_Forward |
5’-ATAGGATGTAATCAGACGACACAGGAA-3’ |
120 |
58.5 |
Exon 16_Reverse |
5’-CACTGCTGGAACTTCGCTCACA-3’ |
120 |
59.4 |
The PCR conditions were an initial denaturation step of 95°C for 10 min, followed by 95°C for 10 s, annealing (annealing temperature specific for a pair of primers) for 15 s and 72°C for 30 s, for a total of 45 cycles. The relative expression levels of APC were normalized to those of actin. The DNA copy number level for the APC exons (exon 5, 9, 12 and 16) in each sample were compared with the level in control blood samples from normal individual. Data were analyzed using the comparative threshold cycle (2–ΔΔCT) method.
ACKNOWLEDGMENTS AND FUNDING
We are grateful to our patients for their sincere participation in this study.
This work was supported by (1) the construction and management of colorectal malignant tumor biological sample bank, Project number 14KG108; (2) FAP new APC mutation family analysis, functional analysis and clinical research, project number 2014KZ058.
CONFLICTS OF INTEREST
The author(s) declare no competing financial interests.
REFERENCES
1. Jiang SS, Li JJ, Li Y, He LJ, Wang QJ, Weng DS, Pan K, Liu Q, Zhao JJ, Pan QZ, Zhang XF, Tang Y, Chen CL, Zhang HX, Xu GL, Zeng YX, Xia JC. A novel pathogenic germline mutation in the adenomatous polyposis coli gene ina Chinese family with familial adenomatous coli. Oncotarget. 2015; 6:27267-74. doi: 10.18632/oncotarget.4776.
2. Song G1, Yuan Y, Zheng F, Yang N. Novel insertion mutation p. Asp610GlyfsX23 in APC gene causes familial adenomatous polyposis in Chinese families. Gene. 2013; 516:204-8.
3. Bulow S. Clinical features in familial polyposis coli. Results of the DanishPolyposisRegister. Dis Colon Rectum 1986; 29:102–7.
4. Jang YH, Lim SB, Kim MJ, Chung HJ, Yoo HW, Byeon JS, Myung SJ, Lee W, Chun S, Min WK. Three novel mutations of the APC gene in Korean patients with familial adenomatous polyposis. Cancer genetics and cytogenetics. 2010; 200:34–39.
5. Aretz S, Hes FJ. Clinical utility gene card for: MUTYHassociated polyposis (MAP), autosomal recessive colorectal adenomatous polyposis. European journal of human genetics. 2010; 18.
6. Aretz S, Genuardi M, Hes FJ. Clinical utility gene card for: MUTYH-associated polyposis (MAP), autosomal recessive colorectal adenomatous polyposis, multiple colorectal adenomas, multiple adenomatous polyps (MAP) - update 2012. European journal of human genetics. 2013; 21.
7. Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D. Identification of FAP locus genes fromchromosome 5q21. Science. 1991; 253:661–5.
8. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, AlbertsenH, Joslyn G, Stevens J, Spirio L, Robertson M. Identification and characterizationof the familial adenomatous polyposis coli gene. Cell. 1991; 66:589–600.
9. Odenwald MA, Prosperi JR, Goss KH. APC/beta-cateninrichcomplexes at membrane protrusions regulate mammarytumor cell migration and mesenchymal morphology. BMCcancer. 2013; 13:12.
10. Ekman M, Mu Y, Lee SY, Edlund S, Kozakai T, Thakur N, Tran H, Qian J, Groeden J, Heldin CH, Landstrom M. APCand Smad7 link TGFbeta type I receptors to the microtubulesystem to promote cell migration. MolBiol Cell. 2012; 23:2109–2121.
11. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988; 319:525-32.
12. Galiatsatos P, Foulkes WD. Familial adenomatouspolyposis. Am J Gastroenterol. 2006; 101:385–398.
13. Aretz S, Uhlhaas S, Sun Y, Pagenstecher C, Mangold E, Caspari R, MosleinG, Schulmann K, Propping P, Friedl W. Familial adenomatous polyposis:Aberrant splicing due to missense or silent mutations in the APC gene. HumMutat. 2004; 24:370-80.
14. Montera M, Piaggio F, Marchese C, Gismondi V, Stella A, Resta N, VarescoL, Guanti G, Mareni C. A silent mutation in exon 14 of the APC gene isassociated with exon skipping in a FAP family. J Med Genet. 2001; 38:863–7.
15. Gismondi V, Bafico A, Biticchi R, Pedemonte S, Di Pietri S, Ponz de Leon M, Groden J, Varesco L. 310 basepair APC deletion with duplication ofbreakpoint (439ins15del310) in an Italian polyposis patient. Hum Mutat. 1998; suppl1:S220–2.
16. Cao X, Eu KW, Seow-Choen F, Zao Y, Cheah PY. APC mutation and phenotypic spectrum of Singapore familial adenomatous polyposis patients. Eur J Hum Genet. 2000; 8:42-8.
17. Petrij-Bosch A, Peelen T, van Vliet M, van Eijk R, Olmer R, DrusedauM, Hogervorst FB, Hageman S, Arts PJ, Ligtenberg MJ, Meijers-HeijboerH, Klijn JG, Vasen HF, Cornelisse CJ, van ’t Veer LJ, Bakker E, van OmmenGJ, Devilee P. BRCA1 genomic deletions are major founder mutations in Dutchbreast cancer patients. Nat Genet. 1997; 17:341–5.
18. Wijnen J, van der Klift H, Vasen H, Khan PM, Menko F, Tops C, MeijersHeijboer H, Lindhout D, Moller P, Fodde R. MSH2 genomic deletions are afrequent cause of HNPCC. Nat Genet. 1998; 20:326–8.
19. Venesio T, Balsamo A, Rondo-Spaudo M, Varesco L, Risio M, Ranzani GN: APC haploinsufficiency, but not CTNNB1 or CDH1gene mutations, accounts for a fraction of familial adenomatouspolyposis patients without APC truncating mutations. Lab Invest. 2003; 83:1859–1866.
20. Liu J, Xing Y, Hinds TR, Zheng J, Xu W. The third 20 amino acidrepeat is the tightest binding site of APC for beta-catenin. J MolBiol 2006; 360:133–144.
21. Rubinfeld B, Souza B, Albert I, Müller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC geneproduct with beta-catenin. Science 1993; 262:1731–1734.
22. Patel BB, Li XM, Dixon MP, Blagoi EL, Nicolas E, SeeholzerSH, Cheng D, He YA, Coudry RA, Howard SD, Riddle DM, Cooper HC, Boman BM, et al. APC +/– alters colonic fibroblast proteome in FAP. Oncotarget. 2011; 2:197–208. doi: 10.18632/oncotarget.241.
23. Kanter-Smoler G, Fritzell K, Rohlin A, Engwall Y, Hallberg B, Bergman A, Meuller J, Grönberg H, Karlsson P, Björk J, Nordling M. Clinical characterization and the mutation spectrum in Swedish adenomatous polyposis families. BMC Med. 2008; 6:10. doi: 10.1186/1741-7015-6-10.
24. Won YJ, Park KJ, Kwon HJ, Lee JH, Kim JH, Kim YJ, Chun SH, Han HJ, Park JG. Germline mutations of the APC gene in Korean familial adenomatous polyposis patients. J Hum Genet. 1999; 44:103-108.
25. Nieuwenhuis MH, VasenHF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev OncolHematol. 2007; 61:153-61.
26. Plawski A, Slomski R. APC gene mutations causing familial adenomatouspolyposis in Polish patients. J Appl Genet 2008; 49:407–414.
27. Caspari R, Friedl W, Mandl M, Möslein G, Kadmon M, Knapp M, Jacobasch KH, Ecker KW, Kreissler-Haag D, Timmermanns G, et al. Familial adenomatous polyposis: mutation atcodon 1309 and early onset of colon cancer. Lancet. 1994; 343:629–632.
28. Rivera B, González S, Sánchez-Tomé E, Blanco I, Mercadillo F, Letón R, Benítez J, Robledo M, Capellá G, Urioste M. Clinical and genetic characterization of classical forms of familial adenomatous polyposis: a Spanish population study. Ann Oncol. 2011; 22:903-9.