
INTRODUCTION
Intracranial germ cell tumors (iGCTs) occur mostly in children and young adults. They account for 3-5% of pediatric CNS tumors with a five- to eightfold higher incidence in East Asia than in Western countries and affect more male than female patients (3:1) [1–3]. iGCTs are classified into pure germinomas and nongerminomatous germ cell tumors (NGGCT), including teratoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma and malignant mixed iGCTs compound of teratomas and one or more of the other histologies. Pure germinoma is the most common subtype followed by mixed iGCTs. An important prognostic factor is the tumor's histology. Pure germinomas are very sensitive to radio- and chemotherapy leading to a 5-year survival rate of more than 90%. In contrast, mixed iGCTs vary in radio- and chemotherapeutic sensitivity; with 5-year survival rates less than 60% [4] for multi-modal therapy. Even though survival rates for germinomas are high long-term outcomes and late effects of radio- and chemotherapy including secondary neoplasms, endocrine deficiencies and strokes, demonstrate that further research with regard to targeted therapy is necessary [5].
The cellular origin of iGCTs is still unclear and controversial. For a long time, a popular hypothesis suggested that all GCTs originate from primordial germ cells (PGCs) migrating in the midline during development. Recent studies argue that iGCTs may develop from neural stem cells [6–8]. In addition, tumor biology underlying the pathogenesis of these tumors is also largely unknown and therefore a subject of current research. However, mutations and other alterations in the KIT/RAS- and AKT/mTOR pathway, as well as gains of chromosomes 12p or X are common biological/genetic alterations in iGCTs and seem to play a crucial role in the tumorigenic process [9–14] although their impact on survival is still unclear.
Gain-of-function-mutations of the tyrosine kinase KIT lead to a permanent pathological activation of this transmembrane protein, independent of binding its ligand stem cell factor (SCF) [15]. This results in an increased and steady stimulation of cell proliferation. A similar mechanism is described for RAS mutations, in particular HRAS, KRAS and NRAS. These small G-proteins play a central part in diverse signalling pathways and are activated by binding GTP. Point mutations in the active center often lead to an inability to bind GTPase which induces stimulation of cell growth [16].
Due to the fact that iGCTs are more frequent in East Asia than in the West, previous studies included mostly Asian patients. Our study is the first genetic analysis of a larger series of germinomas from Caucasian patients. Moreover, we provide immunohistochemical data on global DNA methylation and examined activation of ERK pathway to elaborate further information on the cell of origin, pathogenetic pathway(s) as well as potential novel therapeutic targets.
RESULTS
Clinical features
This study enrolled 55 patients with a median age at diagnosis of 17 years, ranging from 8 to 40 years (Table 1). There were 47 (85.5%) male and 8 (14.5%) female patients. Tumors included in this cohort predominantly affected the region of the pineal gland (31 of 55 [56.4%]) and the suprasellar region (11 of 55 [20%]). 6 germinomas (10.9%) revealed bifocal tumor location, 6 (10.9%) were located in other regions of the brain (pons, hypothalamus and lateral ventricle) and for 1 case (1.8%) information about the exact location was not available. For 33 cases survival data was available, revealing that no patient had metastasis, 6 showed subsequent events and 2 patients died. These 33 patients were all of Caucasian ethnicity. In addition, there was no indication of an Asian or Afroamerican ethnic background from the available data of the other cases. 54 tumors were diagnosed as pure germinomas, only 1 case was considered as mixed GCT with large germinoma contents and a teratoma component. All 55 cases showed c-Kit and Oct4 expression, which are recognized diagnostic criteria for germinomas.
Table 1: Clinical data, genetic alteration status and ERK-/ Akt/mTOR activation in 55 germinoma cases
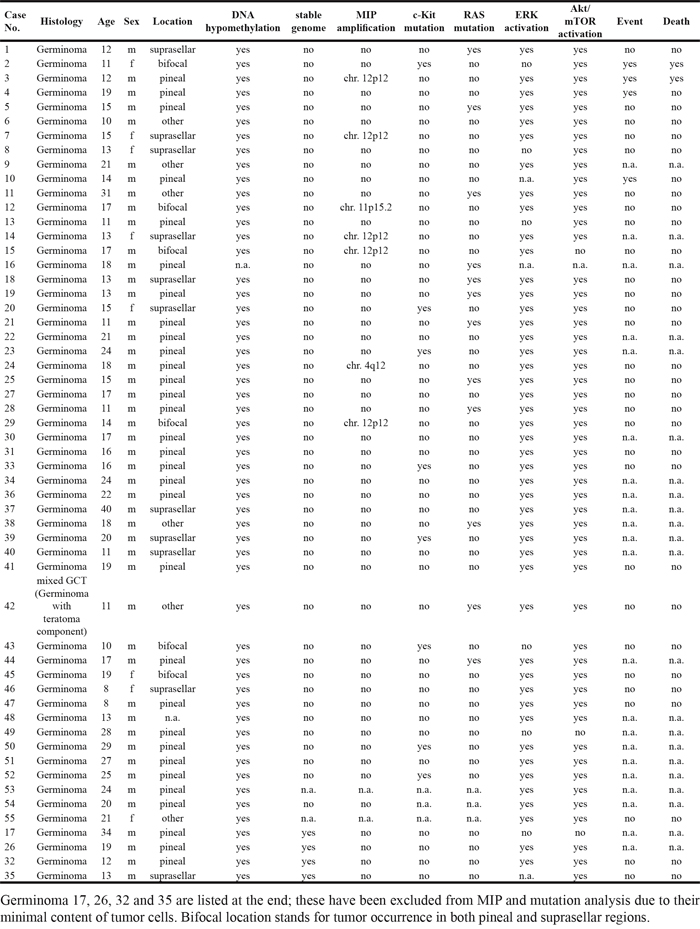
Most interestingly we discovered an association between female gender and age ≤ 15 years (p=0.004) as well as suprasellar tumor location (p=0.016). Conversely, male gender was associated with age >15 years (p=0.004) and pineal tumor location (p=0.023) (Figure 1). Survival data of 33 patients treated according the SIOP/HIT germ cell tumor protocols were available. Progression-free survival or overall survival did not correlate to the genetical or biological alterations evaluated in this study.

Figure 1: Correlation with clinico-pathological data. All p-values refer to the Fisher’s exact test. p-values < 0.05 were considered as statistically significant.
Global hypomethylation of germinomas
Immunohistochemical staining against the 5-Methylcytosine (5mC) epitope of DNA was performed on 54 tumor specimens. All examined samples showed no immunoreactivity for 5mC in tumor cell nuclei, indicating that global DNA hypomethylation is the rule in germinomas (Figure 2). In contrast, peritumoral and tumor-infiltrating lymphocytes, which are characteristic of germinomas, showed strong nuclear staining for this epigenetic hallmark.
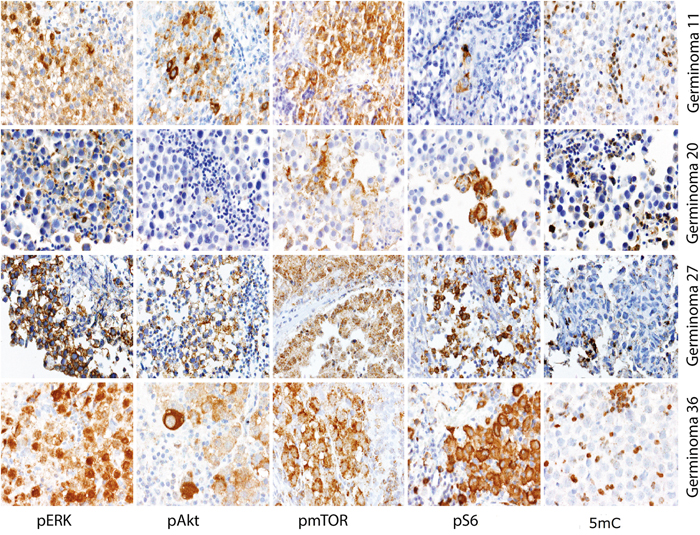
Figure 2: Representative staining results of pERK, pAkt, pmTOR, pS6 and 5mC. All four cases revealed ERK and Akt/mTOR pathway activation and global demethylation.
MIP analysis
Molecular inversion probe analysis was performed for 53 germinomas for which DNA was available. 4 cases (7.5%) showed a stable genome but histologically, they consisted mostly of lymphocytes with only a minimal amount of germinoma cells. For this reason we excluded them from data analysis. We detected extensive genetic alterations and chromosomal instability in all remaining 49 cases (92.5%) (Figure 3). Most frequently, germinomas showed gains on chromosomes 12p (82%), 21q (76%), 8 (67%), 1q (65%) and 7 (59%). Chromosomal losses were less frequent than gains and were most commonly related to chromosomes 13q (45%), 11q (41%), 5q, 9q (both 39%), and 16p (37%).

Figure 3: Virtual karyogram of 49 germinomas included in this study. Chromosomal instability was detected by molecular inversion probe (MIP) arrays. Gains are indicated in blue and losses in red. Frequent gains of chromosomes 1q, 7, 8, 12p and 21q as well as losses of 5q, 9q, 11q, 13q, and 16p become obvious.
Of note, amplification at 12p12, involving KRAS, was detected in 5 cases (10.2%) representing the most frequent amplification. In addition, amplification at 4q12 and 11p15.2, affecting KIT and RRAS-2, were identified in one case (2%) each (Figure 4). Loss of CBL at chromosome 11q23.3 was found in 8 cases (16.3%).
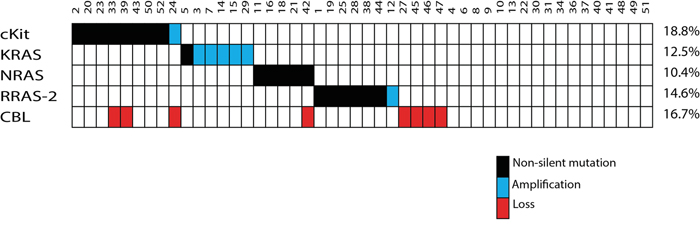
Figure 4: Summary of the somatic events. Each mutation or alteration found in KIT, KRAS, NRAS and RRAS-2 is a mutually exclusive event in the affected germinoma.
GISTIC analysis was used to distinguish significant chromosomal aberrations from random background and revealed a substantial number of copy number (CN) alterations in germinomas. 33 CN gains and 14 CN losses were detected within the germinoma genome by setting the significance cut-off to p≤0.001 (Figure 5). 94% of gains and 79% of losses included protein-coding regions. Remarkably, CN gains affected the IL10 (Interleukin-10) gene and genes encoding its receptors IL10RA, IL10RB and IL10RB-AS1 at chromosomes 1q32.1, 11q23.3 and 21q22.11. Moreover, chromosome 4q12, including KIT, showed also significant CN gains (Table 2).
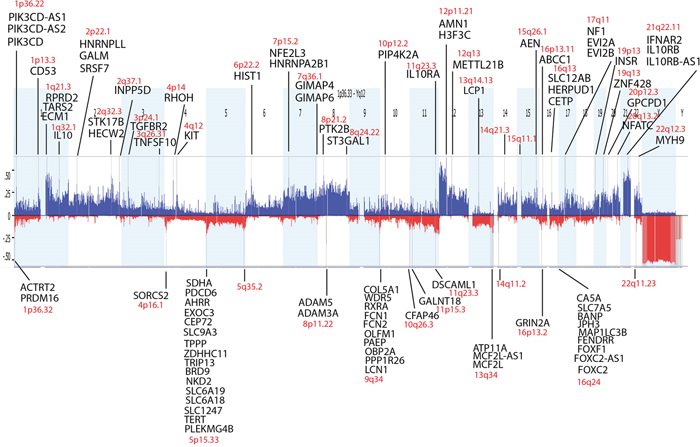
Figure 5: Statistically significant frequent copy number variations in germinomas analyzed by GISTIC. Germinomas reveal a high level of genomic instability. P-values ≤ 0.001 were defined as statistically significant. Protein-coding genes in the affected regions are listed.
Table 2: Protein-coding genes affected by significant CN gains and losses
Region |
Gene |
Name |
|
|---|---|---|---|
Gains |
|||
Chr. 1p36.22 |
PIK3CD-AS1 |
PIK3CD antisense RNA 1 |
|
PIK3CD-AS2 |
PIK3CD antisense RNA 2 |
||
PIK3CD |
Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit delta |
||
Chr. 1p13.3 |
CD53 |
CD53 molecule |
|
Chr. 1q21.3 |
RPRD2 |
Regulation of nuclear pre-mRNA domain containing 2 |
|
TARS2 |
Threonyl-tRNA synthetase 2, mitochondrial (putative) |
||
ECM1 |
Extracellular matrix protein 1 |
||
Chr. 1q32.1 |
IL10 |
Interleukin 10 |
|
Chr. 2p22.1 |
HNRNPLL |
Heterogeneous nuclear ribonucleoprotein L-like |
|
GALM |
Galactose mutarotase (aldose 1-epimerase) |
||
SRSF7 |
Serine/arginine-rich splicing factor 7 |
||
Chr. 2q32.3 |
STK17B |
Serine/threonine kinase 17b |
|
HECW2 |
HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2 |
||
Chr. 2q37.1 |
INPP5D |
Inositol polyphosphate-5-phosphatase D |
|
Chr. 3p24.1 |
TGFBR2 |
Transforming growth factor, beta receptor II (70/80kDa) |
|
Chr. 3q26.31 |
TNFSF10 |
Tumor necrosis factor (ligand) superfamily, member 10 |
|
Chr. 4p14 |
RHOH |
Ras homolog family member H |
|
Chr. 4q12 |
KIT |
v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog |
|
Chr. 6p22.2 |
HIST1 |
Histone cluster 1 |
|
Chr. 7p15.2 |
NFE2L3 |
Nuclear factor, erythroid 2-like 3 |
|
HNRNPA2B1 |
Heterogeneous nuclear ribonucleoprotein A2/B1 |
||
Chr. 7q36.1 |
GIMAP4 |
GTPase, IMAP family member 4 |
|
GIMAP6 |
GTPase, IMAP family member 6 |
||
Chr. 8p21.2 |
PTK2B |
Protein tyrosine kinase 2 beta |
|
Chr. 8q24.22 |
ST3GAL1 |
ST3 beta-galactoside alpha-2,3-sialyltransferase 1 |
|
Chr. 10p12.2 |
PIP4K2A |
Phosphatidylinositol-5-phosphate 4-kinase, type II, alpha |
|
Chr. 11q23.3 |
IL10RA |
Interleukin 10 receptor, alpha |
|
Chr. 12p11.21 |
AMN1 |
Antagonist of mitotic exit network 1 homolog |
|
H3F3C |
H3 histone, family 3C |
||
Chr. 12q13 |
METTL21B |
Methyltransferase like 21B |
|
Chr. 13q14.13 |
LCP1 |
Lymphocyte cytosolic protein 1 (L-plastin) |
|
Chr. 15q26.1 |
AEN |
Apoptosis enhancing nuclease |
|
Chr. 16p13.11 |
ABCC1 |
ATP-binding cassette, sub-family C (CFTR/MRP), member 1 |
|
Chr. 16q13 |
SLC12AB |
Solute carrier family 12 (sodium/chloride transporter), member 3 |
|
HERPUD1 |
Homocysteine-inducible, endoplasmic reticulum stress-inducible, ubiquitin-like domain member 1 |
||
CETP |
Cholesteryl ester transfer protein, plasma |
||
Chr. 17q11 |
NF1 |
Neurofibromin 1 |
|
EVI2A |
Exotropic viral integration site 2A |
||
EVI2B |
Exotropic viral integration site 2B |
||
Chr. 19p13 |
INSR |
Insulin receptor |
|
Chr. 19q13 |
ZNF428 |
Zinc finger protein 428 |
|
Chr. 20p12.3 |
GPCPD1 |
Glycerophosphocholine phosphodiesterase 1 |
|
Chr. 20q13.2 |
NFATC |
Nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 2 |
|
Chr. 21q22.11 |
IFNAR2 |
Interferon (alpha, beta and omega) receptor 2 |
|
IL10RB |
Interleukin 10 receptor, beta |
||
IL10RB-AS1 |
IL10RB antisense RNA 1 (head to head) |
||
Chr. 22q12.3 |
MYH9 |
Myosin, heavy chain 9, non-muscle |
|
Losses |
|||
Chr. 1p36.32 |
ACTRT2 |
Actin-related protein T2 |
|
PRDM16 |
PR domain containing 16 |
||
Chr. 4p16.1 |
SORCS2 |
Sortilin-related VPS10 domain containing receptor 2 |
|
Chr. 5p15.33 |
SDHA |
Succinate dehydrogenase complex, subunit A, flavorprotein (Fp) |
|
PDCD6 |
Programmed cell death 6 |
||
AHRR |
Aryl-hydrocarbon receptor repressor |
||
EXOC3 |
Exocyst complex component 3 |
||
CEP72 |
Centrosomal protein 72kDa |
||
SLC9A3 |
Solute carrier family 9, subfamily A (NHE3, cation proton antiporter 3), member 3 |
||
TPPP |
Tubulin polymerization promotin protein |
||
ZDHHC11 |
Zinc finger, DHHC-type containing 11 |
||
TRIP13 |
Thyroid hormone receptor interactor 13 |
||
BRD9 |
Bromodomain containing 9 |
||
NKD2 |
Naked cuticle homolog 2 (Drosophila) |
||
SLC6A18 |
Solute carrier family 6 (neutral amino acid transporter), member 18 |
||
SLC6A19 |
Solute carrier family 6 (neutral amino acid transporter), member 19 |
||
SLC1247 |
Solute carrier family 12 (potassium/chloride trasporter), member 7 |
||
TERT |
Telomerase reverse transcriptase |
||
PLEKHG4B |
Pleckstrin homology domain containing, family G (with RhoGef domain) member 4B |
||
Chr. 8p11.22 |
ADAM3A |
ADAM metallopeptidase domain 3A (pseudogene) |
|
ADAM5 |
ADAM metallopeptidase domain 5 (pseudogene) |
||
Chr. 9q34 |
COL5A1 |
Collagen, type V, alpha 1 |
|
WDR5 |
WD repeat domain 5 |
||
RXRA |
Retinoid X receptor, alpha |
||
FCN1 |
Ficolin (collagen/fibrinogen domain containing) 1 |
||
FCN2 |
Ficolin (collagen/fibrinogen domain containing lectin) 2 |
||
OLFM1 |
Olfactomedin 1 |
||
PAEP |
Progestogen-associated endometrial protein |
||
OBP2A |
Odorant binding protein 2A |
||
PPP1R26 |
Protein phosphatase 1, regulatory subunit 26 |
||
LCN1 |
Lipocalin 1 |
||
Chr. 10q26.3 |
CFAP46 |
Cilia and flagella associated protein 46 |
|
Chr. 11p15.3 |
GALNT18 |
Polypeptide N-acetylgalactosaminyltransferase 18 |
|
Chr. 11q23.3 |
DSCAML1 |
Down syndrome cell adhesion molecule like 1 |
|
Chr. 13q34 |
ATP11A |
ATPase, class VI, type 11A |
|
MCF2L-AS1 |
MCF2L antisense RNA 1 |
||
MCF2L |
MCF.2 cell line derived transforming sequence-like |
||
Chr. 16p13.2 |
GRIN2A |
Glutamate receptor, ionotropic, N-methyl D-aspartate 2A |
|
Chr. 16q24 |
CA5A |
Carbonic anhydrase VA, mitochondrial |
|
SLC7A5 |
Solute carrier family 7 (amino acid transporter light chain, L system), member 5 |
||
BANP |
BTG3 associated nuclear protein |
||
JPH3 |
Junctophilin 3 |
||
MAP1LC3B |
Microtubule-associated protein 1 light chain 3 beta |
||
FENDRR |
FOXF1 adjacent non-coding developmental regulatory RNA |
||
FOXF1 |
Forkhead box F1 |
||
FOXC2-AS1 |
FOXC2 antisense RNA 1 |
Molecular analysis of c-Kit and RAS mutations in germinomas
We examined a total of 51 germinomas and 1 mixed GCT (germinoma and teratoma component) for mutations in KIT exons 11, 13, 17 and 18 as well as mutation hotspots in HRAS, KRAS, NRAS and RRAS-2. For the 4 cases (7.7%) showing a stable genome in MIP analysis, no mutations could be detected in KIT or RAS. Due to their minute amount of tumor cells they were excluded from the sequence data analysis.
Sanger sequencing exposed non-silent KIT mutations in 8 cases (16.7%) (Figure 6). Most mutations affected tyrosine kinase II region (TK2) encoded by exon 17 in terms of point mutations in codons 816 (3/52) and 820 (2/52). In addition, one deletion of codon 560 in exon 11 and 2 point mutations in codon 634 of exon 13 were detected (representative sequencing results of KIT mutations are given in Figure 7a). No mutation in exon 18 of the KIT was observed.
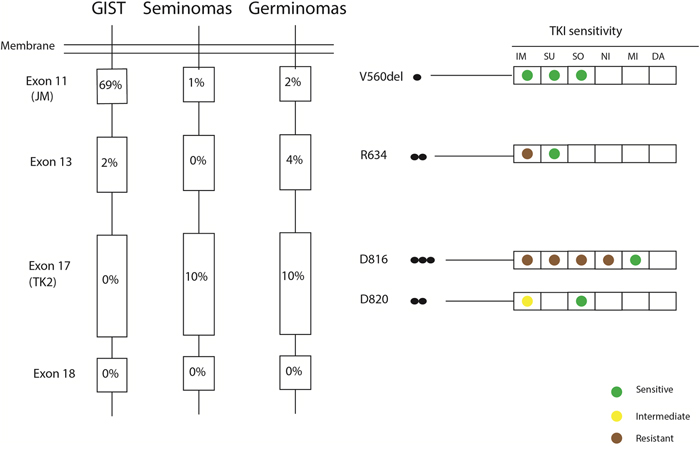
Figure 6: Somatic KIT mutations found in this germinoma cohort in comparison to reported KIT mutations in gastrointestinal stromal tumors (GISTs) and seminomas. Black circles represent the number of cases harboring a given mutation. The functional domains concerned by mutations are juxtamembrane domain (JM) and tyrosine kinase II (TK2). Previously described tyrosine-kinase inhibitors (TKIs) [imatinib (IM), sunitinib (SU), sorafenib (SO), nilotinib (NI), midostaurin (MI) and dasatanib (DA)] and there activity against each mutation are shown on the right.
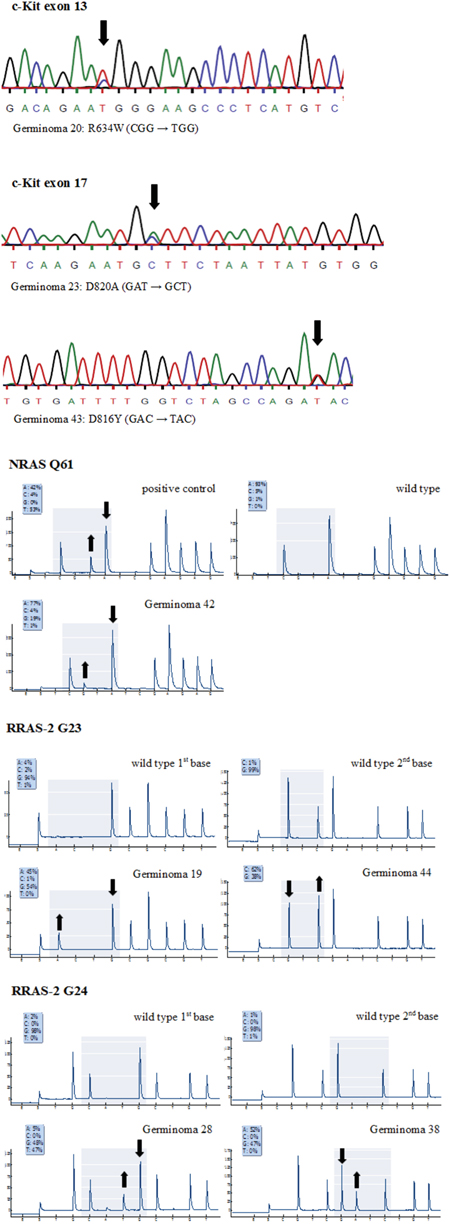
Figure 7: a. Representative KIT mutations detected by Sanger sequencing. b. Representative cases of RAS mutations detected by pyrosequencing analysis. Pyrograms are compared to wild type and/or positive control data. Significant peak increases and concomitant reductions in germinoma 42 [NRAS Q61R (CAA → CGA)], 19 [RRAS-2 G23S (GGC → AGC)], 44 [RRAS-2 G23A (GGC → GCC)], 28 [RRAS-2 G24C (GGC → TGC)] and 38 [RRAS-2 G24D (GGC → GAC)] expose mutations in these cases.
Pyrosequencing analysis of the RAS mutation hotspots codon G12, G13 and Q61 in HRAS, KRAS and NRAS as well as their homologous parts codon G23, G24 and Q72 in RRAS-2, revealed 12 (25%) non-silent point mutations. Mainly, these point mutations affected the GTP binding domain (Figure 8).
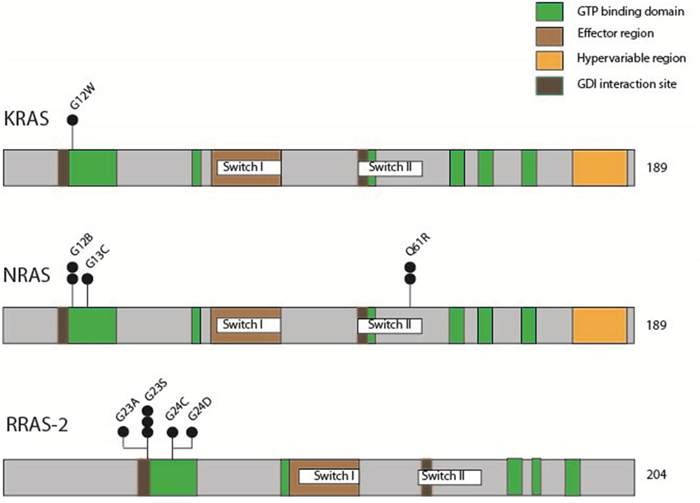
Figure 8: Schematic representation of mutations in KRAS, NRAS and RRAS-2, illustrating involvement of functional gene domains. Black circles indicate the number of mutations identified.
Only one point mutation was seen in KRAS affecting codon G12. NRAS G12 and Q61 were each mutated in 2 tumors and NRAS G13 in 1. Most remarkably, no specimen revealed mutations in HRAS, but 6 (12.5%) germinomas showed point mutations in the RAS homolog RRAS-2 of which 4 involved codon G23 and 2 involved codon G24 (representative sequencing results of RAS mutations are given in Figure 7b).
Altogether, genetic alterations were observed in 27 cases (56.3%) in KIT or RAS genes which were mutually exclusive (Figure 4). Comparison of KIT/RAS mutation status in germinomas and patient's age, sex and tumor location revealed no significant correlations (Figure 1).
Immunohistochemical analysis of ERK- and Akt/mTOR-pathway
Immunohistochemical staining against pAkt, pmTOR, pS6 and pERK was performed on 54 GCTs including 53 pure germinomas and 1 mixed GCT (germinoma and teratoma component). Nuclear and cytoplasm staining of these proteins was considered positive.
pERK expression was observed in 46 (88.5%) tumors. Expression scores ranged from 0 to 300 (median, 102). 10 (19.2%) tumor samples showed strong staining for pERK. 24 (46.2%) tumor specimen revealed moderate staining whereas in 12 (23%) cases weak staining was found. No immunoreactivity for pERK was detected in 6 cases (11.5%).
45 (84.9%) tumor specimens showed expression of pAKT. Expression scores ranged from 0 to 300 (median, 101). Strong staining for pAkt was observed in 7 cases (13.2%). We detected moderate staining in 30 (56.6%) and weak staining in 8 (15.1%) samples. Negative staining was seen in 8 (15.1%) tumors.
pmTOR expression was detected in 45 (90%) tumor specimens. Expression scores ranged from 0 to 270 (median, 87). 3 (6%) cases were considered as strongly positive. 40 (80%) samples were rated moderate whereas 2 (4%) tumors disclosed weak staining. 5 (10%) germinomas revealed no immunoreactivity for the used pmTOR antibody.
In contrast to the other stainings, staining for pS6 was generally negative [31 tumors (57.4%)]. Only 23 (42.6%) samples revealed immunoreactivity for pS6. Expression scores ranged from 0 to 200 (median, 33). Strong staining was seen in only one (1.9%) case. 14 (25.9%) tumors showed moderate and 8 (14.8%) exhibited weak staining (Figure 9).
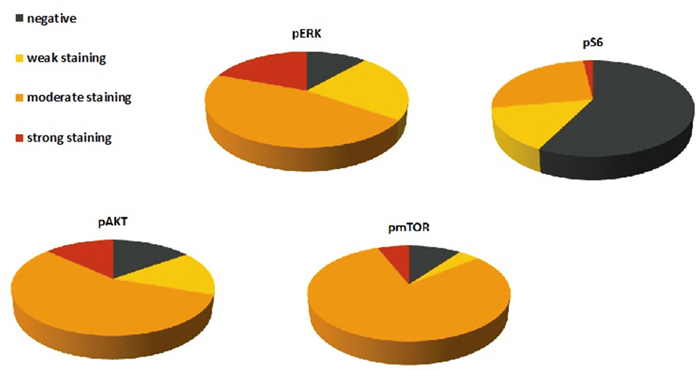
Figure 9: Protein expression of pERK, pAkt, pmTOR and pS6 in 54 germinomas. Graphic presentation of distribution of negative, weak, moderate and strong staining against pERK, pAkt, pmTOR and pS6. pERK, pAkt and pmTOR were frequently expressed whereas pS6 showed more negative results.
In seven cases, immunostaining could not be assessed because the samples contained only scant tumor cells in the presence of many lymphocytes (1 case for pAkt, 2 cases for pERK and 4 cases for pmTOR).
In summary, 88.5% of germinomas revealed an upregulation of the ERK pathway. Activation of AKT/mTOR pathway was seen in 94.4% of germinomas (illustration of IHC scores in Figure 10). Most remarkably, activation of both pathways was detected in 83.3% (representative staining results of cases with activations in both signal pathways are given in Figure 2).
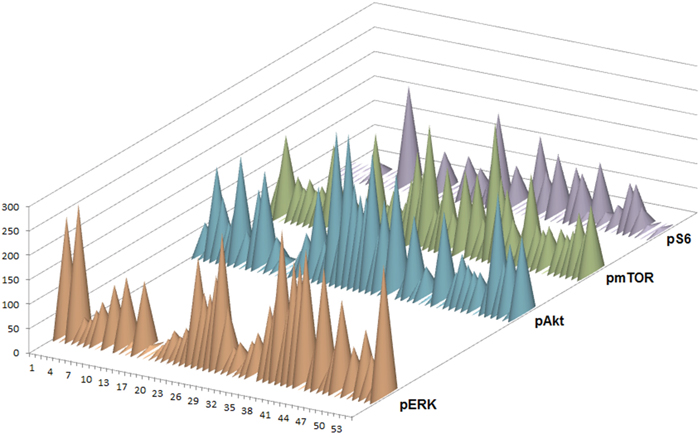
Figure 10: Semiquantitive analysis of pERK, pAkt, pmTOR and pS6 expression by IHC in 54 germinomas. Staining was scored by multiplying intensity by percentage of stained cells. The result was a score between 0 and 300.
DISCUSSION
Our knowledge about the cellular origin of iGCTs is based on a limited number of studies. Analysis on mouse embryos showed that PGCs physiologically migrate in midline body structures during development, finally residing in the genital ridges [17]. Germ cells that remain in other areas usually die through reduction of stem cell factor signaling which leads to apoptosis of these PGCs [18]. In addition, Hoei-Hansen et al. [6] showed expression of genes associated with embryonic stem cell pluripotency like PLAP, HCG, OCT-3/4, KIT, AP-2y and NANOG in iGCTs. One hypothesis on the cellular origin of iGCTs is that they are derived from mismigrated PGCs. Alternatively, Tan and Scotting [7] suggested that transformation of endogenous neural stem cells by demethylation of the OCT-4 promotor results in an upregulation of this gene which might trigger the formation of GCTs. Moreover, they assume that gene-specific methylation and additional genetic events, like KIT mutations in germinomas, might play a role in specification of these tumors. However, this hypothesis raises the question on the triggering mechanism involved in the demethylation of neural stem cells. In addition, this model does not explain why expression of c-Kit is not restricted to pure germinomas but also found as component in mixed GCTs [9].
Considering human germ cell development, global hypomethylation is an epigenetic hallmark in primordial germ cells (PGCs) accompanied by high c-Kit expression [19, 20]. We examined a large cohort of germinomas and showed that 100% of these revealed global hypomethylation and c-Kit expression in immunohistochemical staining, independently of c-Kit mutational status. In contrast to germinomas, studies revealed global methylation for yolk sac tumors [21] and non-seminomas [22], indicating a fundamental difference with germinomas in terms of lineage differentiation or cell of origin. Other brain tumor entities including different glioma entities, medulloblastomas and rhabdoid tumors of the CNS have been extensively characterised in the last years by genome-wide methylation profiling and did not show a similar demethylation phenotype [23–25]. Comparing the KIT mutational frequencies of exons 11, 13, 17 and 18 encoding different functional domains between germinomas, seminomas and gastrointestinal germ cell tumors (GISTs) showed strong similarities between seminomas and germinomas but a different distribution for GISTs [26, 27]. These results reinforce the genetic similitudes for seminomas and germinomas. Broader information on methylation status and c-Kit expression in larger collectives of NGGCTs could further help understanding differences of the pathogenesis in subtypes of iGCTs. Global DNA demethylation demonstrated by immunohistochemistry for 5mC may be a useful tool for the differential diagnosis of iGCTs. In addition, differences in therapeutic response between pure germinomas and NGGCTs could also be related to the tumors' methylation status.
In our study we showed that global hypomethylation of tumor DNA strikingly coincides with chromosomal instability in germinomas. Previously, Fukushima et al. [9] reported frequent gains of chromosomes 21q (66.7%), 1q (56.7%) and X (53.5%) and loss of chromosome 13q (53.3%) in a larger cohort of germinomas. These findings were confirmed by Wang et al. [14], describing chromosomes X (72.7%) and 21q (63.6%) as frequently amplified and 11q (45.5%), 13q and 17p (both 36.4%) frequently deleted in germinomas. Our findings reinforce these discoveries, showing frequent gains on chromosomes 12p (82%), 21q (76%), 8 (67%), 1q (65%) and 7 (59%) and losses on chromosomes 13q (45%), 11q (41%), 5q, 9q (both 39%), and 16p (37%) in pure germinomas. Of note, chromosome 12p is frequently affected by chromosomal gains in our study. In addition, amplification of 12p12, involving the KRAS gene, was detected in 10% of cases. This leads us to assume that amplification of 12p seems to play an important role in germinomas. In the last years, several reports already highlighted the importance of chromosome 12p in testicular GCTs, especially of amplification at 12p11.2-p12.1 [28] and describing it as a genetic hallmark of germ cell tumors [29]. Our results strongly confirm these findings for germinomas. On the other hand, previous studies reported the frequency of 12p gain in germinomas as only 36.7% [9] and 36.4% [14], which is a smaller percentage than in our cohort. We propose three explanations for this finding. First, we examined a larger number of germinomas than in the previous studies and maybe therefore obtained more representative data. Second, we examined only tumors of Caucasian patients whereas the previous studies mostly investigated Asian patients. Geographical genetic differences might explain the results. Third, Fukushima et al. [9] described 10% of germinomas as chromosomally stable. In our initial screening, we also identified 4 germinomas of 53 (7.5%) which showed a stable genome in MIP analysis. However, these tumors had a minute amount of tumor cells (< 5%) and mainly consisted of lymphocyte infiltrates contrary to the other samples. For this reason, we suspect that allele frequencies of these tumors are too low to be detected as genetic alterations by MIP analysis. This is also corroborated by the fact that these 4 tumors did not reveal any alterations in the mutation analysis (see below). GISTIC analysis revealed significant CN gains in the IL10 gene and its receptor genes. Sredni et al. [30] showed that autocrine/paracrine IL10 secretion of stomach adenocarcinoma and glioblastoma multiforme cell lines is essential for tumor cell proliferation and inhibition of IL10 leads to decreased cell proliferation. Future studies might show if germinoma proliferation also depends on IL10.
MIP analysis also detected amplifications at 4q12 and 11p15.2, affecting the KIT and RRAS-2 loci in one case each. As described before, physiological primordial germ cell growth is dependent on KIT expression and iGCTs show pathological activation of c-KIT and downstream signaling components. For this reason, we statistically examined the KIT and RAS family member mutational status in our cohort. We found genetic alterations in 17.3% of germinomas for KIT and 34.6% for RAS whereas Fukushima et al. [9] detected c-Kit mutations in 40% and RAS mutations in 20%, Wang et al. [14] 25% for KIT and 19% for RAS and recently Ichimura et al. [31] found c-Kit mutated in 40% and RAS mutated in 19% of germinomas. In summary, our overall fraction of alterations in KIT and RAS is similar to the previous studies. However we found a higher percentage of alterations in RAS compared to KIT. This difference in the distribution between our cohort and the three collectives above might be due to the ethnicity of the patients leading to geographically different frequencies of genetic alterations in germinomas. Remarkably, all alterations in KIT and RAS were mutually exclusive genetic events in our cohort. Fukushima et al. [9], Wang et al. [14] and Ichimura et al. [31], who found 97% of germinomas having mutually exclusive mutations in KIT or RAS, confirmed this finding, which strongly suggests that an alteration in one signal component is sufficient to trigger tumorigenesis. Moreover, in our series we identified RRAS-2 as a new player in germinoma pathogenesis, which we found alterated in 14.6% of cases.
Furthermore, we discovered activation of the Akt/mTOR pathway by immunohistochemistry in 94.4% and upregulation of ERK pathway in 88.5% of germinomas. Activation of both pathways was detected in 83.3%. Interestingly, pS6 was found to be negative in 57.4% of cases indiacting that this component is not the preferentially phosphorylated target of mTOR in germinomas. Indeed, in other cell types such as cerebellar granule neuron precursors it was shown that mTOR is able to induce eIF4E, which is essential for proliferation, and simultaneously suppresses S6 activity, which is needed for cell cycle exit [32]. Therefore, S6 is not necessary for cell proliferation in cerebellar granule neuron precursors. The main mTOR targets in germinomas and their functional roles have to be identified in future studies.
These findings indicate that activation of Akt/mTOR and ERK pathways are general phenomena in germinomas. Therefore, we suspect that the 43.7% of germinomas with no genetic alterations in our study might have alteration in other components of these pathways besides KIT and RAS. This is supported by Ichimura et al. [31] who found 7% of germinomas mutated for MTOR and further tumors mutated in genes of these two pathways.
Current therapy for intracranial germinomas achieves excellent long term survival, albeit at the cost of some morbidity. Targeted inhibition of the ERK and Akt/mTOR pathways might be an option for patients with recurrent tumors of whose tumors fail to respond to first line therapy. Recently, small molecule inhibitors of Akt were tested at different levels of clinical trials [33]. Phase 1 clinical trials showed positive results for the oral Akt inhibitor afuresertib [34]. In addition, in vitro studies with the ERK inhibitor SCH772984 in RAS mutant cells exhibited promising results [35]. Ichimura et al. [31] showed that mTor mutant cells which were treated with the mTor inhibitor pp242 showed dose-dependent downregulation of the Akt/mTor pathway. Combining ERK and Akt/mTOR inhibitors might be a therapeutic option for patients with recurrences that could be related to over compensatory hyperactivation of the parallel signal pathway, when only one pathway was inhibited before. However, the dependence of tumor cells on these pathways has to be proven when tumor models become available and further investigations are required to examine targeted therapy options for germinomas.
MATERIALS AND METHODS
Tumor specimen
The study includes 54 histologically verified pure germinomas and 1 mixed intracranial germ cell tumor which contained large germinoma parts and some teratoma components. Tumor specimens were retrieved from the archives of the Institute of Neuropathology, University of Bonn Medical Center (Bonn, Germany), of the DGNN German Brain Tumor Reference Center (Bonn, Germany) and the German Paediatric Tumor Registry (Kiel, Germany). All tumors were diagnosed according to the WHO classification of tumors of the central nervous system [36] using histological and immunohistochemical methods.
DNA extraction
Hematotoxylin-eosin (H&E) –stained sections of each case were examined carefully before DNA extraction. DNA from 53 formalin-fixed paraffin-embedded (FFPE) tumors was extracted using the QIAamp DNA Mini Tissue Kit (Qiagen GmbH; Düsseldorf, Germany) according to the manufacturer’s protocol. All tissue samples were used in an anonymous manner, as approved by the ethics committee at the University of Bonn Medical Center (Bonn, Germany).
Molecular inversion profiling
To identify copy number gains and losses, we used an MIP array using OncoScan FFPE Express 330K Platform Version 2 (Affymetrix, Santa Clara, CA, USA). The molecular inversion probe assay of 53 germinoma samples was performed as already described [37]. By using Nexus Copy Number 7.0 Discovery Edition software (BioDiscovery, El Segundo, CA, USA) the raw MIP data file was analyzed. To make copy number and loss of heterozygosity calls, BioDiscovery’s SNP-FASST2-Segmentation algorithm was used. Genomic Identification of Significant Targets in Cancer (GISTIC) analysis was used to distinguish significant chromosomal aberrations from random background [38].
KIT mutation analysis
52 tumors were screened for mutations in KIT exons 11, 13, 17 and 18 by direct sequencing. Therefore, polymerase-chain-reaction-products were purified using a PCR purification kit (Qiagen) and sent to a commercial sequencing service (Eurofins MWG Operon, Ebersberg, Germany), where direct sequencing of DNA was performed in duplicate (forward and reverse) using 30 ng of PCR products.
Pyrosequencing analysis for mutation hot spots of NRAS, KRAS, HRAS and RRAS-2
52 specimens were analyzed for hot spot mutations in the RAS genes NRAS, KRAS, HRAS and RRAS-2. PCR amplification primers flank the homologous hotspot regions of these genes. For the pyrosequencing reaction, single-stranded DNA templates were immobilized on streptavidin-coated Sepharose high-performance beads (GE Healthcare, Uppsala, Sweden) using the PSQ Vacuum Prep Tool and Vacuum Prep Worktable (Biotage, Uppsala, Sweden), according to the manufacturer’s instructions. Then DNA was incubated at 80°C for two minutes and allowed to anneal to 0.4 mmol/L sequencing primer at room temperature. Pyrosequencing was performed using PyroGold Reagents according to the manufacturer’s protocol. Positive and negative controls were used to compare results. Pyrograms were analyzed by PyroMark Q24 software (Biotage), using the allele quantification (AQ) software to determine the percentage of mutant versus wild type alleles according to percentage relative peak height. A sequential nucleotide dispensation protocol was used, reflecting the expected order of nucleotide incorporation and the potential base change within the first, second or third position of the hot spot codons G12, G13 and Q61 of HRAS, NRAS and KRAS. In the same manner codons G23, G24 and Q72 of RRAS-2 were examined. Peak heights are proportional to the number of nucleotides that are incorporated with each dispensation.
Immunohistochemistry
Paraffin-embedded tissue samples were divided into serial sections of 4 μm. Positively charged slides were cut, air dried overnight at 37°C, deparaffinized in xylene and rehydrated in a degraded alcohol sequence. Slides were then microwaved in 10 mmol/L citrate buffer (pH 6.0) for antigen retrieval, followed by incubation in 3% hydrogen peroxide for 5 minutes at room temperature to block the activity of endogenous peroxidase and in blocking solution (CSA II Kit; Dako, Glostrup, Denmark). Tumors were incubated overnight with primary antibodies [pAkt Ser473, 1:200, rabbit monoclonal antibody (736E11, Cell Signaling Technology, Leiden, Netherlands); pERK Thr202/Tyr204, 1:200, rabbit monoclonal antibody (20G11, Cell Signaling Technology); pS6 Ser235/236, 1:200, rabbit monoclonal antibody (91B2, Cell Signaling Technology); pmTOR Ser2448, 1:50, rabbit monoclonal antibody (49F9, Cell Signaling Technology); 5-Methylcytosine, 1:8000, mouse monoclonal antibody (BI-MECY, IgG1, Eurogentec, Seraing, Belgium)]. Sections were washed in Tris-buffered saline with Tween 20. Visualization of bound antibodies was achieved by the CSA system embodies technology (CSA II, Biotin-Free catalyzed Amplification System; Dako). Slides were counterstained with hematoxylin and dehydrated in a graded alcohol sequence and mounted in Richard-Allan Scientific Cytoseal XYL (Thermo Scientific, Waltham, MA).
Two authors blinded for clinical and genomic data evaluated the percentage of stained cells and scored the samples for staining intensity (0 = negative, 1 = weak intensity, 2 = moderate intensity, 3 = strong intensity). The results were used to calculate a score between 0 and 300 by multiplying the percentage of stained cells by intensity, and tumor samples were classified into negative staining (score 0-9), weak staining (score 10-49), moderate staining (score 50-199) and strong staining (score 200-300).
Statistical analysis
Significantly overrepresented factor values in a particular factor group were identified using the one-tailed Fisher’s Exact test at a p-value of 0.05. The log-rank test statistic [39] was used to identify regions yielding a high degree of survival prediction. p-values were calculated based on a Chi-Squared test and p-value ≤ 0.05 were considered significant. To compare survival times, Kaplan-Meier curves were generated. The p-values were computed using the log-rank test [39].
ACKNOWLEDGMENTS
We thank Dr. Steffen Albrecht, Montreal, for critical reading of the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Echevarria ME, Fangusaro J, Goldman S. Pediatric central nervous system germ cell tumors: a review. Oncologist. 2008; 13:690-699.
2. Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol. 2013; 15:ii1-56.
3. Packer RJ, Cohen BH, Cooney K. Intracranial germ cell tumors. Oncologist. 2000; 5:312-320.
4. Matsutani M, Sano K, Takakura K, Fujimaki T, Nakamura O, Funata N, Seto T. Primary intracranial germ cell tumors: a clinical analysis of 153 histologically verified cases. J Neurosurg. 1997; 86:446-455.
5. Acharya S, DeWees T, Shinohara ET, Perkins SM. Long-term outcomes and late effects for childhood and young adulthood intracranial germinomas. Neuro Oncol. 2015; 17:741-746.
6. Hoei-Hansen CE, Sehested A, Juhler M, Lau YF, Skakkebaek NE, Laursen H, Rajpert-de Meyts E. New evidence for the origin of intracranial germ cell tumours from primordial germ cells: expression of pluripotency and cell differentiation markers. J Pathol. 2006; 209:25-33.
7. Tan C, Scotting PJ. Stem cell research points the way to the cell of origin for intracranial germ cell tumours. J Pathol. 2013; 229:4-11.
8. Teilum G. Classification of endodermal sinus tumour (mesoblatoma vitellinum) and so- called “embryonal carcinoma” of the ovary. Acta Pathol Microbiol Scand. 1965; 64:407-429.
9. Fukushima S, Otsuka A, Suzuki T, Yanagisawa T, Mishima K, Mukasa A, Saito N, Kumabe T, Kanamori M, Tominaga T, Narita Y, Shibui S, Kato M, et al. Mutually exclusive mutations of KIT and RAS are associated with KIT mRNA expression and chromosomal instability in primary intracranial pure germinomas. Acta Neuropathol. 2014; 127:911-925.
10. Kamakura Y, Hasegawa M, Minamoto T, Yamashita J, Fujisawa H. C-kit gene mutation: common and widely distributed in intracranial germinomas. J Neurosurg. 2006; 104:173-180.
11. Okada Y, Nishikawa R, Matsutani M, Louis DN. Hypomethylated X chromosome gain and rare isochromosome 12p in diverse intracranial germ cell tumors. J Neuropathol Exp Neurol. 2002; 61:531-538.
12. Sakuma Y, Sakurai S, Oguni S, Satoh M, Hironaka M, Saito K. c-kit gene mutations in intracranial germinomas. Cancer Sci. 2004; 95:716-720.
13. Schneider DT, Zahn S, Sievers S, Alemazkour K, Reifenberger G, Wiestler OD, Calaminus G, Gobel U, Perlman EJ. Molecular genetic analysis of central nervous system germ cell tumors with comparative genomic hybridization. Mod Pathol. 2006; 19:864-873.
14. Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, Nakamura H, He Z, Doddapaneni H, Lewis L, Wang M, Suzuki T, Nishikawa R, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature. 2014; 511:241-245.
15. Tian Q, Frierson HF, Jr., Krystal GW and Moskaluk CA. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol. 1999; 154:1643-1647.
16. Goodsell DS. The molecular perspective: the ras oncogene. Oncologist. 1999; 4:263-264.
17. Molyneaux KA, Stallock J, Schaible K, Wylie C. Time-lapse analysis of living mouse germ cell migration. Dev Biol. 2001; 240:488-498.
18. Runyan C, Schaible K, Molyneaux K, Wang Z, Levin L, Wylie C. Steel factor controls midline cell death of primordial germ cells and is essential for their normal proliferation and migration. Development. 2006; 133:4861-4869.
19. Gkountela S, Li Z, Vincent JJ, Zhang KX, Chen A, Pellegrini M, Clark AT. The ontogeny of cKIT+ human primordial germ cells proves to be a resource for human germ line reprogramming, imprint erasure and in vitro differentiation. Nat Cell Biol. 2013; 15:113-122.
20. Smallwood SA, Kelsey G. De novo DNA methylation: a germ cell perspective. Trends Genet. 2012; 28:33-42.
21. Jeyapalan JN, Noor DA, Lee SH, Tan CL, Appleby VA, Kilday JP, Palmer RD, Schwalbe EC, Clifford SC, Walker DA, Murray MJ, Coleman N, Nicholson JC, Scotting PJ. Methylator phenotype of malignant germ cell tumours in children identifies strong candidates for chemotherapy resistance. Br J Cancer. 2011; 105:575-585.
22. Rijlaarsdam MA, Tax DM, Gillis AJ, Dorssers LC, Koestler DC, de Ridder J, Looijenga LH. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS One. 2015; 10:e0122146.
23. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012; 22:425-37.
24. Pietsch T, Schmidt R, Remke M, Korshunov A, Hovestadt V, Jones DT, Felsberg J, Kaulich K, Goschzik T, Kool M, Northcott PA, von Hoff K, von Bueren AO, et al. Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol. 2014; 128:137-49.
25. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, Jones DTW, Sturm D, Hermann C, Wang MS, Korshunov A, Rhyzova M, Grobner S, et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell. 2016; 29:379-393.
26. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011; 11:865-878.
27. Coffey J, Linger R, Pugh J, Dudakia D, Sokal M, Easton DF, Bishop DT, Stratton M, Huddart R, Rapley EA. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: Report of 220 tumors and review of literature. Gene Chromosome Canc. 2008; 47:34-42.
28. Rodriguez S, Jafer O, Goker H, Summersgill BM, Zafarana G, Gillis AJ, van Gurp RJ, Oosterhuis JW, Lu YJ, Huddart R, Cooper CS, Clark J, Looijenga LH, Shipley JM. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene. 2003; 22:1880-1891.
29. Cheng L, Zhang S, Eble JN, Beck SD, Foster RS, Wang M, Ulbright TM. Molecular genetic evidence supporting the neoplastic nature of fibrous stroma in testicular teratoma. Mod Pathol. 2012; 25:1432-1438.
30. Sredni B, Weil M, Khomenok G, Lebenthal I, Teitz S, Mardor Y, Ram Z, Orenstein A, Kershenovich A, Michowiz S, Cohen YI, Rappaport ZH, Freidkin I, Albeck M, Longo DL, Kalechman Y. Ammonium trichloro(dioxoethylene-o,o')tellurate (AS101) sensitizes tumors to chemotherapy by inhibiting the tumor interleukin 10 autocrine loop. Cancer Res. 2004; 64:1843-1852.
31. Ichimura K, Fukushima S, Totoki Y, Matsushita Y, Otsuka A, Tomiyama A, Niwa T, Takami H, Nakamura T, Suzuki T, Fukuoka K, Yanagisawa T, Mishima K, et al. Recurrent neomorphic mutations of MTOR in central nervous system and testicular germ cell tumors may be targeted for therapy. Acta Neuropathol. 2016; 131:889-901.
32. Mainwaring LA, Kenney AM. Divergent functions for eIF4E and S6 kinase by sonic hedgehog mitogenic signaling in the developing cerebellum. Oncogene. 2011; 30:1784-1797.
33. Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010; 19:1355-1366.
34. Spencer A, Yoon SS, Harrison SJ, Morris SR, Smith DA, Brigandi RA, Gauvin J, Kumar R, Opalinska JB, Chen C. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood. 2014; 124:2190-2195.
35. Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, Long B, Liu J, Dinunzio E, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013; 3:742-750.
36. Louis DN, Cavenee WK, Wiestler OD, Ohgaki H, eds. WHO classification of tumours of the central nervous system. IARC, Lyon 2007.
37. Wang Y, Cottman M, Schiffman JD. Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer Genet. 2012; 205:341-355.
38. Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, Du J, Kau T, Thomas RK, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007; 104:20007-20012.
39. Peto R, Peto J. Asymptotically Efficient Rank Invariant Test Procedures. J R Stat Soc Ser a-G. 1972; 135:185.