
INTRODUCTION
The histological and molecular classification of breast cancer (BC) has drastically helped to characterize and treat patients, although the clinical resolution is an enigma. A limited number of biomarkers can be used to manage BC patients and the estrogen receptor alpha (ESR1) is essential for hormonal therapy. Unfortunately, almost 30% of BCs are ESR1 negative or acquire resistance to hormonal therapy [1,2]. Among specific biomarkers that predict the response to therapy or can be utilized as new therapeutic targets, pRb is a strong candidate [2].
The pRb pathway is frequently inactivated in breast cancer via a phosphorylation dependent mechanism that is driven by the overexpression of cyclin D1 or inactivation of the CDK-inhibitor p16ink4a. pRb gene or protein loss has also been reported at different frequencies [3]. Deletion of Rb1 in mouse mammary stem/bipotent progenitor cells induced focal acinar hyperplasia with squamous metaplasia that progressed in transplantable mammary tumors similar to either luminal-B or TNT subtypes [4].
pRb is a multifaceted tumor suppressor protein that controls many pathways but has been well-described only in its role in cell cycle control. pRb acts as a repressor of the cell cycle by inhibiting the activity of E2F transcription factors. Hyper-phosphorylated pRb releases E2F transcription factors and allows for the expression of genes that mediate entry into the S phase [5]. Emerging evidence suggests that pRb has a more complex role in cancer initiation and progression [6] and understanding which functional biological nodes are altered in pRb negative cells is an important question in order to realize personalized therapy.
There is a convincing association between ESR1 and pRb status. Histological analyses of different breast cancer subtypes showed a prevalence of pRb loss in ESR1 negative tumors [7] with a high frequency of occurrence in triple-negative breast cancer subtypes [8]. From a therapeutic point of view, a gene expression signature of pRb-dysfunction is associated with a relatively poor response to endocrine therapy and a better prognosis following chemotherapy treatment that is widely utilized in the treatment of ER-negative disease [3,82013;10].
These data suggest an interaction between the pRb pathway and the status of ESR1. In turn, there is some evidence in the literature that has driven us to hypothesize a direct link between the pRb and ESR1 protein functionality. pRb is a cofactor for more than a hundred proteins [6] including nuclear receptors. Rb indirectly enhances the activity of glucocorticoid receptors and inhibits the thyroid hormone receptor and PPARgamma-dependent transcription [11]. In cancer cells, pRb modulates the activity of the AR and ESR1, the two principal determinants of hormonal cancer. pRb interacts with the androgen receptor in a hormone-independent manner (Lu and Danielsen jbc 1998) [12] and can regulate its activity via the E2F transcription factor 1 resulting in a critical determinant of therapeutic response [13]. Finally, pRb interacts in a ligand-dependent manner with the RIZ protein, an ESR1 cofactor that can also potentiate SRC-2 activity on ESR1 signaling [11]. Conversely, the direct activity of pRb on ESR1 protein function remains largely unknown.
Here we show that the retinoblastoma protein (pRb) is fundamental for ESR1 basal turnover and activity. We demonstrated that loss of pRb in human breast cancer cells or human/mouse primary mammary cells, but not the two related family members p107 and pRb2/p130, decreases the expression of the ESR1 protein. Treatment with proteasome inhibitors re-establishes the expression of ESR1 demonstrating the involvement of the proteasome pathway. As confirmed, in RB1 knock down cells, ESR1 is ubiquitinated to a greater degree than in normal cells. Mechanistically, the N-terminal domain of pRb interacts with the CD domain of ESR1 to allow for the interaction of chaperone proteins and in particular, HSP90 and p23. We demonstrated that pRb is important for the formation of a chaperone intermediate complex on ESR1.
RESULTS
pRb controls the ESR1 protein level and activity
To test our hypothesis, we have generated MCF7 (ESR1 positive) cell lines knocked-down for the three members of the Retinoblastoma family, pRb, pRb2/p130, and p107 (Supplemental Figure 1A) [14,15]. Supplemental Figure 1B shows that the loss of pRb family members decreased the expression of ESR1 when compared to scrambled cells. Among the three members, only pRb is involved in this mechanism (Supplemental Figure 1B, Figure 1A). The data were obtained in basal conditions in the absence of hormones (Charcoal Stripped Serum, CSS). We decided to perform all the experiments under these conditions unless otherwise indicated. To exclude that the mechanism is a characteristic of a single cell line, we have down regulated pRb in the T47D ESR1 positive breast cancer cell line. The results in T47D cells are comparable to those in the MCF7 cells (Figure 1C). In both cell lines, the downregulation of ESR1 in RB1 kd cells is statistically significant (Figure 1B,D). To confirm the data, we have carried out immunofluorescence experiments. We observed a reduction in signal intensity of the ESR1 in MCF7 RB1 kd cells in basal and estradiol-stimulated conditions (Figure 1E). To definitively demonstrate that the activity of ESR1 was compromised, we have assessed the expression of some classical ESR1 target genes [16]. We observed that the expression of TFF1 and CTSD are down regulated in RB1 kd cells (Supplemental Figure 1C). Analysis of ESR1 mRNA also showed a reduction in RB1 kd cells (Figure 2A). Since the pRb family members could bind the ESR1 promoter [17] and the ESR1 protein itself regulates its expression [16], we have cloned the ESR1 downstream a non-endogenous promoter. A western blot analysis indicates that the reduction of ESR1 relative expression under the non-endogenous promoter is comparable with that of endogenous promoter indicating a control at the posttranscriptional level (Figure 2B). These data indicate that pRb could be a new cofactor of ESR1, regulating its protein expression level.
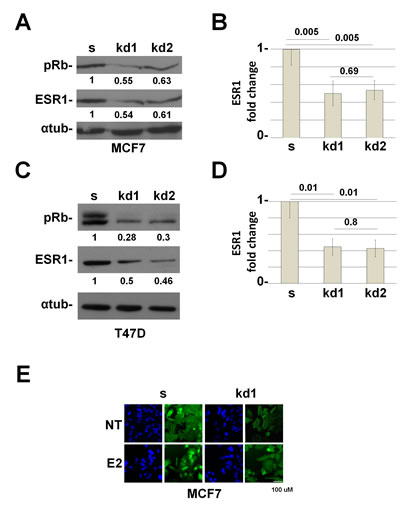
Figure 1: RB1 kd MCF7 and T47D cells down regulate the ESR1 protein. (A) ESR1 expression in RB1 knocked down MCF7 cells. kd1 and kd2 represent two different shRNAs. Alpha-tubulin was used as a loading control. (B) Quantification of three independent experiments as in (A). y axis represents the ratio between the ESR1 and alpha-tubulin proteins and normalized to scrambled cells. The RB1 knocked down MCF7 cells express about 50% less of the ESR1 protein. (C) ESR1 expression in RB1 knock down T47D cells. (D) Quantification of three independent experiments as in (b). The RB1 knock down T47D cells express about 56% less of the ESR1 protein. (E) MCF7 cells were grown in CSS medium for 3 days (NT) and treated with 10-8 estradiol for 45 minutes (E2). Cells were probed with an ESR1 antibody (green) and stained with DAPI (blue). RB1 kd1 cells express less ESR1 protein.
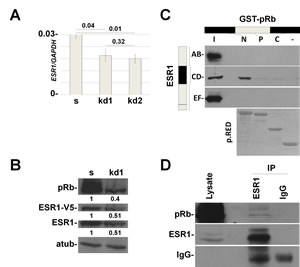
Figure 2: In vitro and in vivo interaction between pRb and ESR1 proteins. (A) RB1 kd1 and kd2 MCF7 cell lines have about 30% less mRNA levels of ESR1. y axis represents the ratio between ESR1 and GAPDH mRNAs. (B) MCF7 cells were infected with a lentivirus that expresses ESR1-V5 under the CMV promoter. RB1 kd1 cells express less endogenous and transfected ESR1 protein, demonstrating that the effect is at the posttranslational level. Quantification represents the ratio between the indicated protein and alpha-tubulin and normalized to scrambled cells. (C) In vitro interaction. ESR1 AB, CD and EF domains were pulleddown with GST-pRb N-terminal, Pocket and C-terminal domains in MCF7 cells. There is an interaction between the ESR1CD and pRb N-terminal domains. p.RED, pounceau red. (D) In vivo interaction. ESR1 was immunoprecipitated in MCF7 cells and analyzed by western blot with a pRb antibody. IgG was used as a negative control.
pRb and ESR1 form a protein complex
To test a possible interaction between pRb and ESR1, we have carried out a GST pull down assay with the three functional domains of pRb [5,18] and the AB, CD and EF domains of the ESR1 protein in MCF7 cells [19]. As highlighted in Figure 2C, the pRb N-terminal domain interacts with the CD domain of ESR1. To definitively demonstrate this interaction, we have performed a co-immunoprecipitation assay on endogenous proteins. Figure 2D shows the in vivo interaction between pRb and ESR1.
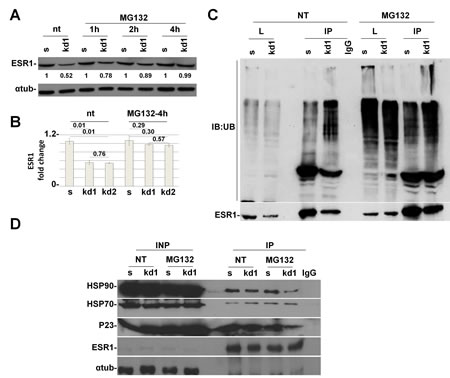
Figure 3: pRb is necessary for HSP90 protein to protect ESR1 from proteasomal degradation. (A) Time course analysis of MCF7 cells treated with MG132 for indicated time. After 4 hours, the RB1 kd1 cells expressed the same level of ESR1 as scrambled cells. (B) Analysis as in (A) of three independent experiments utilizing RB1 kd1 and kd2 shRNAs. (C) MCF7 cell lysate untreated (nt) or treated with MG132 for 4 hours was immunoprecipitated with an ESR1 antibody and analyzed by western blot with an anti-ubiquitin antibody. The RB1 kd1 cells showed heavy ubiquitination that extended in the stacking gel. (D) MCF7 cell lysate untreated (nt) or treated with MG132 for 4 hours was immunoprecipitated with ESR1 antibody. In RB1 kd1 cells, there is less HSP90 and p23 proteins bound to the ESR1 protein. HSP70 protein is unaltered. IgG was used as a negative control.
pRb controls the basal turnover of ESR1 via the proteasome pathway
ESR1, as with most of the hormonal receptors, is finely regulated at the transcriptional and posttranscriptional levels. A key role in protein half-life is played by the proteasome pathway [192013;21]. Without the hormone, the ESR1 is associated with HSP70, HSP40 and the adapter HIP protein (HSP70-interacting protein) to form an early complex. Later on, HSP90 and the adapter protein HOP (HSP70/HSP90- organizing protein) displace HSP40 and bind the hydrophobic hormone-binding domain of ESR1 to form an intermediate complex [22]. After ATP binding, HSP90 interacts with p23 and Cyclophilin 40 (CYP40) to form a mature complex [22]. To test if pRb is involved in the ESR1 degradation via the proteasome pathway, we have treated the MCF7 cells with the proteasome inhibitor MG132 [23]. After 4 hours of treatment, RB1 kd cells had the same level of ESR1 as scrambled cells, thus rescuing the phenotype observed in untreated cells (Figure 3A,B). This finding was confirmed in RB1 kd2 cells (Supplemental Figure 2A) and utilizing the drug Bortezomib (Supplemental Figure 2B), another proteasome inhibitor. The same results were also observed in T47D cells (Supplemental Figure 2C). To assess if ESR1 is more ubiquitinated in RB1 kd MCF7 cells, we treated the sample with MG132, immunoprecipitated with an ESR1 antibody and analyzed the ubiquitin level. Figure 3C confirmed that ESR1 is more ubiquitinated in RB1 kd cells as compared to scrambled cells. A typical smear is observed in the stacking gel. As a consequence of the obtained results, we have analyzed the chaperone proteins involved in ESR1 protein stability [24]. In Figure 3D, we show the co-immunoprecipitaion of ESR1 with HSP90. It appears that RB1 kd1 cells have less HSP90 bound to ESR1 in untreated and MG132-treated samples compared to scrambled cells. The HSP90 co-chaperone, p23, is also reduced as expected. The level of HSP70 is unaltered, suggesting that pRb influences the intermediate complex that stabilizes the ESR1 protein [22]. Under basal conditions, the ESR1 is associated with chaperone proteins in the cytoplasm. After estradiol stimulation, ESR1 is shuttled to the nucleus to exert its genomic function on target genes [25]. The re-cycling of ESR1 on promoters of target genes is very fast [26], after which the ESR1 is exported again to the cytoplasm for proteasomal degradation [23]. Inhibitors of nuclear export such as Leptomycin B [27] or, nuclear stabilizing agents such as cycloheximide lead to ESR1 protein accumulation. When RB1 kd1 cells were treated with Leptomycin B (Figure 4A) or cycloheximide (Figure 4B) in combination with estradiol, the level of ESR1 were comparable with scrambled cells, confirming a cytoplasmic ESR1 degradation driven by the proteasome pathway.
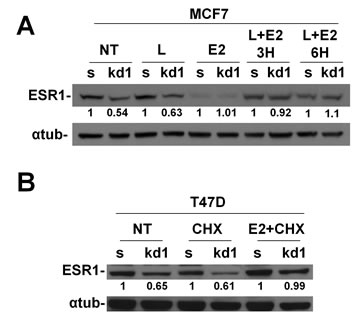
Figure 4: Nuclear accumulation of ESR1 prevents proteasomal degradation in RB1 kd1 cells. (A) MCF7 cells were treated with leptomycin B, estradiol or a combination for indicated time. Estradiol allows the translocation of ESR1 in the nucleus and Leptomycin B prevents the nuclear export. After 3 or 6 hours of treatment, the RB1 kd1 cells express the same level of ESR1 as scrambled cells. (B) T47D cells were treated with CHX, estradiol, or a combination for 3 hours. CHX allows nuclear accumulation of ESR1. After CHX plus estradiol treatment, the RB1 kd1 cells express the same level of ESR1 as scrambled cells.
pRb controls ESR1 protein levels in primary human mammary cells and Rb1 KO mice.
To assess if the mechanism takes place in a more physiological model, we have knocked-down RB1 in human mammary epithelial primary cells (HMEpC). In these cells, we were able to detect the short isoform of ESR1 [28]. In Figure 5A, we show that RB1 kd cells have a reduction in ESR1 levels when compared to scrambled cells and MG132 can rescue the phenotype as in MCF7 and T47D cancer cell lines. To definitively demonstrate the role of pRb in the control of ESR1 protein levels, we have conditionally knocked out (KO) the Rb1 gene in the mouse mammary epithelium. Western blot analysis shows that the expression of the pRb protein is reduced by 50% in KO mice when compared to wild type mice (Figure 5B,C).
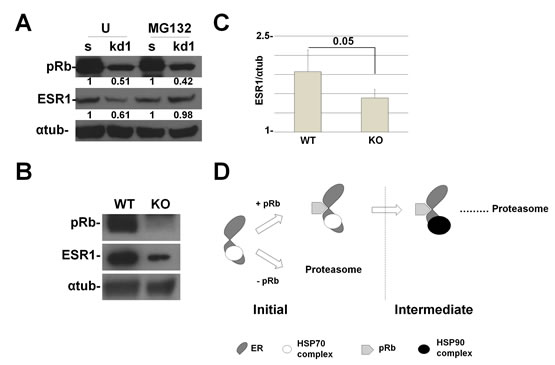
Figure 5: RB1 kd primary human mammary cells and Rb1 KO mice show a reduction in ESR1 protein levels. (A) HMEpC RB1 kd1 cells express 50% less of the ESR1 protein compared to scrambled cells. Treatment with MG132 for 4 hours rescued the expression of the ESR1 protein. The quantification represents the ratio between the ESR1 and alpha-tubulin proteins and normalized to scrambled cells. (B) Rb1 conditional KO mice in mammary cells have a reduced expression of ESR1. (C) Quantification of 6 mice as in (B). (D) Working model of pRb and the chaperone complex that regulate ESR1 turnover. After the assembly of the initiation complex, pRb is necessary for the recruitment of the HSP90 complex that forms the intermediate complex. Without pRb, the ESR1 is more ubiquitinated and degraded by proteasome pathway.
Discussion
Our data here show that the pRb protein plays an important role as an adaptor in the regulation of ESR1 stability and functionality, a key player in hormonal therapy. The N-terminal domain of pRb interacts with the ESR1 CD domain, which is composed of the DNA binding domain (C) and the hinge region (D). The hinge region has recently been discovered to have a complex function. This domain contains a nuclear translocation signal and includes estrogen-independent regulatory sequences, which mediate the interaction with c-Jun and Sp-1 transcription factors [29]. In addition, the domain is modified by many posttranslational modifications [30,31] and together with the ligand binding domain form a surface for HSP90 binding which protects unliganded ESR1 from basal turnover [32]. We anticipate that pRb represents a new actor for the CD domain, which is fundamental for the binding of HSP90, thus regulating the fine equilibrium of rapid switch on/off that characterizes the activity of ESR1 protein. It is interesting to note that the N-terminal domain of pRb mediates this interaction. Although most of pRb’s interactions were mapped in the pocket domain, recently the N-terminal domain has gained much attention. It is involved in inhibiting the E2F transactivation ability [33], can interact with different proteins that control DNA replication [34], and form a complex with SRC-2, a coactivator of nuclear receptors including ESR1 [11]. There is a growing body of evidence that the N-terminal domain may have a role in tumor suppression [6,35,36] and our work highlights its importance in the regulation of ESR1 activity with a direct consequence in the response of breast cancer therapy.
Finally, previous papers have shown the pRb can act as cofactor to control the stability of different target proteins [6,362013;38]. Interactions between pRb and the RIM domain of E2F1 or with PDX-1, a transcription factor involved in pancreas development and adult β-cell functions, protects these proteins from the ubiquitin-proteasome pathway degradation [362013;38]. Our model suggests a role for pRb as a cofactor that allows the interaction between HSP90 and ESR1 to form the chaperone intermediate complex. In the absence of pRb, the formation of the intermediate complex does not occur, thus priming the ESR1 for proteasomal degradation (Figure 5D). In accordance with the literature, blocking HSP90 function with geldanamycin disrupts the interaction with the ESR1 and promotes it’s degradation [39].
During tumor evolution, most hormonal-dependent cancers lack hormone responsiveness and dependence. Since steroid hormones induce differentiation, one explanation is that uncontrolled tumor proliferation is incompatible with differentiation. In this context, our data suggest the pRb loss, a central player of cellular proliferation and differentiation, dictates the steroid response and tumor growth. Current breast cancer therapy is based on a few molecular targets [402013;44] and it has been challenging to find new candidates thus far. pRb loss can define a class of breast tumors that would experience little benefit from endocrine therapy [2,45]. Therapies that aim to reactivate the function of the pRb protein [9] can help those patients that are not responsive to hormonal therapy.
Methods
Cells culture conditions
MCF7 and T47D breast cancer cell lines were purchased from American Type Culture Collection, Rochville, MD, USA, 293FT cell lines were from Invitrogen Carlsbad, CA, USA and HMEpC from Cell Application, San Diego, CA, USA. Cells were grown at 37 0C, in a 5% CO2/95% atmosphere according to the manufacturer’s guidelines. Hormone-free medium was prepared with phenol red–free DMEM with 2 mmol/L L-glutamine, 0.1 mmol/L nonessential amino acids, 50 units/mL penicillin, 50 µg/mL streptomycin, and 3% charcoal-stripped FBS.
Reagents
Antibodies for the following proteins: ERα (sc-8002), ERα HC-20 (sc-543), ERα MC-20 (sc-542), pRb (sc-102), Hsp70 (sc-24) and Hsp90 (sc-69703) were purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA; α-tubulin (T-6074) from Sigma Inc., St Louis, MO, USA; P23 (MA3414) from Thermo Scientific Pierce; and anti-ubiquitin P4DI from Covance, Princeton, NJ, USA. MG-132 (10012628) from Cayman Chemical; Bortezomib (B-1408) and Leptomycin B (L-6100) from LC Laboratories; Cycloheximide (c4859) from Sigma; and (Z)-4-Hydroxytamoxifen from Enzo Life Sciences.
Plasmids
shRNA plasmids RB1 (TRCN0000010418, TRCN0000040167) were from Sigma Inc., St Louis, MO, USA. Scrambled shRNA (Addgene: 17920), psPAX2 packaging plasmid (Addgene: 12260), and pMDG.2 envelop plasmid (Addgene: 12259) were from Addgene Inc, Cambridge, MA, USA. ERα- His-AB, His-CD and His-EF plasmids were derived from VP16-ERα (Addgene: 11351) following amplification with primers: AB-BamHI-F ATG GAT CCA CCA TGA CCA TGA CCC TCC-3’, AB-EcoRI-Rev 5’-ATG AAT TCT CCT TGG CAG ATT CCA TAG-3’, CD-BamHI-F 5’-ATG GAT CCA CCA TGG CCA AGG AGA CTC GCT ACT GTG-3’, CD-EcoRI-Rev 5’-ATG AAT TCT TCT TAG AGC GTT TGA TCA TG-3’. EF-BamHI-F 5’-ATG GAT CCA CCA TGT CTA AGA AGA ACA GCC TGG CC-3’, EF-EcoRI-Rev 5’-ATG AAT TCC AGA CCG TGG CAG GGA AAC-3’. After BamHI/EcoRI double digestion, fragments were ligated into a pcDNA6 His/Myc vector. To generate the ERα-V5 plasmid, we have recombined the pLOVE vector (Addgene:15948) and the pENTR223- ERα, (DF-HCC: HSCD00376961) utilizing the Gateway Cloning System (Invitrogen). GST-Rb N-terminal (1-373 a.a.), Pocket (379-792a.a.) and C-terminal (793-920 a.a.) were cloned in PGEX-2T. All the plasmids were sequence verified.
Lentiviral production
To generate knock down cells, lentiviral particles were produced as described (http://www.broadinstitute.org/genome_bio/trc/publicProtocols.html) by Rizzolio et. al [5].
Real-time PCR
Total RNA was prepared from tissues using the RNA extraction kit RNAeasy (Qiagen Inc, Valencia, CA, USA). One μg of total RNA was reverse transcribed in a 20 μl reaction using M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Primers to amplify ER, CTDS, TFF1, GAPDH are the following: ESR1-for 5’ -CAT TCT ACA GGC CAA ATT CAG - 3’, ESR1-rev 5’ -GCA CAC TGC ACA GTA GCG A - 3’, CTDS-f 5’-GCT GGG AGG CAA AGG CTA CAA-3’ CTDS-r 5’-TCC TGC TCT GGG ACT CTC CT-3’, TFF1-f 5’-CCC TGG TGC TTC TAT CCT AAT A-3’, TFF1-r 5’-AGA AGC GTG TCT GAG GTG TCC-3’, GAPDH-f 5-GAA GGT GAA GGT CGG AGT-3’, GAPDH-r 5-CAT GGG TGG AAT CAT ATT GGA-3’. Quantitative Real Time PCR (qRT-PCR) was performed with SYBR Green PCR Master Mix (Roche Diagnostic, Basel, Switzerland) in a LightCycler® 480 Real-Time PCR System instrument (Roche Diagnostic, Basel, Switzerland). Samples were run in triplicates and the efficiency of each primer was calculated utilizing an internal standard control [46]. All values were normalized for GAPDH.
Co-immunoprecipitation assay
Sub-confluent MCF7 cells were harvested and proteins were prepared as follows: the cell pellet was resuspended in lysis buffer (20 mM Tris HCl pH 8, 137 mM NaCl, 10% glycerol, 1% NP40, 2 mM EDTA). 3 mg of proteins was immunoprecipitated, utilizing 4 μg of ERα antibody or mouse IgG overnight at 4°C. Extracts were incubated with antibodies and protein A/G beads (Pierce) for 3 h at 4°C. Immunopellets were washed extensively and subjected to SDS-PAGE followed by immunoblot analyses to detect pRb and ERα proteins.
Immunofluorescence
MCF7 scramble and pRb KD cells were seeded on cover slips and grown in hormone-free medium for three days. Cells were treated with 17-β-estradiol or ethanol (as a negative control) for 45 minutes and fixed in 4% paraformaldehyde, permeabilized with phosphate-buffered saline (PBS) containing 1% Triton X-100 and 1% bovine serum albumin, and blocked in blocking buffer (PBS containing 8% bovine serum albumin). Cells were then incubated with total ERα antibody diluted at 1:100 (HC-20). After three washing with PBS, Alexa Fluor dyes (Invitrogen) were applied in blocking buffer as a secondary antibody. Nuclei were stained with 2’,6’-diamidino-2-phenylindole (DAPI) in an antifade mounting medium (Vector laboratories, Burlingame, Calif.).
Mice
All experiments with mice were approved by Temple IACUC committee. pRb floxed (FVB;129) and WAP-CRE (B6.Cg) mice were obtained from MMHCC (Mouse Models of Human Cancers Consortium). Mammary tissue was obtained from age-, parous- matched female mice, one day post-partum. Tissues were homogenized in Lysis Buffer (20 mM Tris HCl pH 8, 137 mM NaCl, 10% glycerol, 1% NP40, 2 mM EDTA) and 5-10 ug of protein extract was analysed by western blot.
Statistical analysis
Statistical analysis was performed using GraphPad software by applying Student’s t-test.
ACKNOWLEDGEMENTS
We thank Mr. Robert Fratamico for his assistance in copyediting the manuscript. European Research Council, Programme “Ideas”, Proposal No 269051 (G.T., F.R.), Associazione Italiana per la Ricerca sul Cancro (AIRC) (A.G.) and Special Program Molecular Clinical Oncology, 561000, n°12214) (G.T.), Italian Ministry of Education MIUR (FIRB prot. RBAP11ETKA (I.C., G.T.)), Istituto Tumori Toscano (A.G.), Sbarro Health Research Organization (www.shro.org) (A.G.), Human Health Foundation Spoleto-Terni (www.hhfonlus.org) (A.G.) Commonwealth of Pennsylvania (A.G.). The authors declare no conflict of interest.
References
1. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nature reviews. Cancer. 2009 Sep;9(9):631–43.
2. Bosco EE, Wang Y, Xu H, Zilfou JT, Knudsen KE, Aronow BJ, Lowe SW, Knudsen ES. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. The Journal of clinical investigation. 2007 Jan;117(1):218–28.
3. Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, Knudsen ES. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell cycle. 2010 Oct 15;9(20):4153–63.
4. Jiang Z, Deng T, Jones R, Li H, Herschkowitz JI, Liu JC, Weigman VJ, Tsao M-S, Lane TF, Perou CM, Zacksenhaus E. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. The Journal of clinical investigation. 2010 Sep;120(9):3296–309.
5. Rizzolio F, Lucchetti C, Caligiuri I, Marchesi I, Caputo M, Klein-Szanto AJ, Bagella L, Castronovo M, Giordano A. Retinoblastoma tumor-suppressor protein phosphorylation and inactivation depend on direct interaction with Pin1. Cell Death Differ. 2012 Jul;19(7):1152–61.
6. Viatour P, Sage J. Newly identified aspects of tumor suppression by RB. Dis Model Mech. 2011;4(5):581–5.
7. Derenzini M, Donati G, Mazzini G, Montanaro L, Vici M, Ceccarelli C, Santini D, Taffurelli M, Treré D. Loss of retinoblastoma tumor suppressor protein makes human breast cancer cells more sensitive to antimetabolite exposure. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008 Apr 1;14(7):2199–209.
8. Treré D, Brighenti E, Donati G, Ceccarelli C, Santini D, Taffurelli M, Montanaro L, Derenzini M. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Annals of oncology. 2009 Nov;20(11):1818–23.
9. Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010 Jul 15;29(28):4018–32.
10. Stengel KR, Dean JL, Seeley SL, Mayhew CN, Knudsen ES. RB status governs differential sensitivity to cytotoxic and molecularly-targeted therapeutic agents. Cell cycle. 2008 Apr 15;7(8):1095–103.
11. Batsché E, Desroches J, Bilodeau S, Gauthier Y, Drouin J. Rb enhances p160/SRC coactivator-dependent activity of nuclear receptors and hormone responsiveness. The Journal of biological chemistry. 2005 May 20;280(20):19746–56.
12. Yeh S, Miyamoto H, Nishimura K, Kang H, Ludlow J, Hsiao P, Wang C, Su C, Chang C. Retinoblastoma, a tumor suppressor, is a coactivator for the androgen receptor in human prostate cancer DU145 cells. Biochemical and biophysical research communications. 1998 Jul 20;248(2):361–7.
13. Sharma A, Comstock CE, Knudsen ES, Cao KH, Hess-Wilson JK, Morey LM, Barrera J, Knudsen KE. Retinoblastoma tumor suppressor status is a critical determinant of therapeutic response in prostate cancer cells. Cancer Res. 2007;67(13):6192–203.
14. Rizzolio F, Esposito L, Muresu D, Fratamico R, Jaraha R, Caprioli GV, Giordano A. RB gene family: genome-wide ChIP approaches could open undiscovered roads. J Cell Biochem. 2010 Apr 1;109(5):839–43.
15. Caligiuri I, Rizzolio F, Boffo S, Giordano A, Toffoli G. Critical choices for modeling breast cancer in transgenic mouse models. J Cell Physiol. 2011 Aug;227(8):2988–91.
16. Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38(11):1289–97.
17. Macaluso M, Montanari M, Noto PB, Gregorio V, Bronner C, Giordano A. Epigenetic modulation of estrogen receptor-alpha by pRb family proteins: a novel mechanism in breast cancer. Cancer research. 2007 Aug 15;67(16):7731–7.
18. Rizzolio F, Caligiuri I, Lucchetti C, Fratamico R, Tomei V, Gallo G, Agelan A, Ferrari G, Toffoli G, Klein-Szanto AJ, Giordano A. Dissecting Pin1 and phospho-pRb regulation. J Cell Physiol. 2012 May 2;
19. Leclercq G, Lacroix M, Laïos I, Laurent G. Estrogen receptor alpha: impact of ligands on intracellular shuttling and turnover rate in breast cancer cells. Current cancer drug targets. 2006 Feb;6(1):39–64.
20. Sherman MY. Proteotoxic stress targeted therapy (PSTT). Oncotarget. 2011 May;2(5):356–7.
21. Hildenbrand ZL, Molugu SK, Herrera N, Ramirez C, Xiao C, Bernal RA. Hsp90 can accommodate the simultaneous binding of the FKBP52 and HOP proteins. Oncotarget. 2(1-2):43–58.
22. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nature reviews. Cancer. 2005 Oct;5(10):761–72.
23. Laïos I, Journé F, Nonclercq D, Vidal DS, Toillon R-A, Laurent G, Leclercq G. Role of the proteasome in the regulation of estrogen receptor alpha turnover and function in MCF-7 breast carcinoma cells. The Journal of steroid biochemistry and molecular biology. 2005 Mar;94(4):347–59.
24. Tao YJ, Zheng W. Chaperones and the maturation of steroid hormone receptor complexes. Oncotarget. 2011 Mar;2(3):104–6.
25. Green KA, Carroll JS. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nature reviews. Cancer. 2007 Sep;7(9):713–22.
26. Reid G, Hübner MR, Métivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Molecular cell. 2003 Mar;11(3):695–707.
27. Nonclercq D, Journé F, Laïos I, Chaboteaux C, Toillon R-A, Leclercq G, Laurent G. Effect of nuclear export inhibition on estrogen receptor regulation in breast cancer cells. Journal of molecular endocrinology. 2007 Aug;39(2):105–18.
28. Kang KS, Morita I, Cruz A, Jeon YJ, Trosko JE, Chang CC. Expression of estrogen receptors in a normal human breast epithelial cell type with luminal and stem cell characteristics and its neoplastically transformed cell lines. Carcinogenesis. 1997 Feb;18(2):251–7.
29. Burns KA, Li Y, Arao Y, Petrovich RM, Korach KS. Selective mutations in estrogen receptor alpha D-domain alters nuclear translocation and non-estrogen response element gene regulatory mechanisms. The Journal of biological chemistry. 2011 Apr 8;286(14):12640–9.
30. Atsriku C, Britton DJ, Held JM, Schilling B, Scott GK, Gibson BW, Benz CC, Baldwin MA. Systematic mapping of posttranslational modifications in human estrogen receptor-alpha with emphasis on novel phosphorylation sites. Mol Cell Proteomics. 2009;8(3):467–80.
31. Sentis S, Le Romancer M, Bianchin C, Rostan MC, Corbo L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol Endocrinol. 2005;19(11):2671–84.
32. Berry NB, Fan M, Nephew KP. Estrogen receptor-alpha hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Molecular endocrinology. 2008 Jul;22(7):1535–51.
33. Burke JR, Deshong AJ, Pelton JG, Rubin SM. Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. The Journal of biological chemistry. 2010 May 21;285(21):16286–93.
34. Ahlander J, Chen X-B, Bosco G. The N-terminal domain of the Drosophila retinoblastoma protein Rbf1 interacts with ORC and associates with chromatin in an E2F independent manner. PloS one. 2008 Jan;3(7):e2831.
35. Yang H, Williams BO, Hinds PW, Shih TS, Jacks T, Bronson RT, Livingston DM. Tumor suppression by a severely truncated species of retinoblastoma protein. Molecular and cellular biology. 2002 May;22(9):3103–10.
36. Campanero MR, Flemington EK. Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proceedings of the National Academy of Sciences of the United States of America. 1997 Mar 18;94(6):2221–6.
37. Hofmann F, Martelli F, Livingston DM, Wang Z. The retinoblastoma gene product protects E2F-1 from degradation by the ubiquitin-proteasome pathway. Genes & development. 1996 Dec 1;10(23):2949–59.
38. Kim Y-C, Kim SY, Mellado-Gil JM, Yadav H, Neidermyer W, Kamaraju AK, Rane SG. RB regulates pancreas development by stabilizing Pdx1. The EMBO journal. 2011 Apr 20;30(8):1563–76.
39. Lee MO, Kim EO, Kwon HJ, Kim YM, Kang HJ, Kang H, Lee JE. Radicicol represses the transcriptional function of the estrogen receptor by suppressing the stabilization of the receptor by heat shock protein 90. Molecular and cellular endocrinology. 2002 Feb 25;188(1-2):47–54.
40. Harris TJR, McCormick F. The molecular pathology of cancer. Nature reviews. Clinical oncology. 2010 May;7(5):251–65.
41. Hammond MEH, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. Journal of clinical oncology. 2010 Jun 1;28(16):2784–95.
42. Bosco EE, Knudsen ES. RB in breast cancer: at the crossroads of tumorigenesis and treatment. Cell cycle. 2007 Mar 15;6(6):667–71.
43. Subjeck JR, Repasky EA. Heat shock proteins and cancer therapy: the trail grows hotter! Oncotarget. 2011 Jun;2(6):433–4.
44. Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011 Jun;2(6):435–47.
45. Witkiewicz AK, Ertel A, McFalls J, Valsecchi ME, Schwartz G, Knudsen ES. RB-Pathway Disruption Is Associated with Improved Response to Neoadjuvant Chemotherapy in Breast Cancer. Clinical cancer research. 2012 Sep 15;18(18):5110–22.
46. Rizzolio F, Bione S, Sala C, Tribioli C, Ciccone R, Zuffardi O, Di Iorgi N, Maghnie M, Toniolo D. Highly conserved non-coding sequences and the 18q critical region for short stature: a common mechanism of disease? PLoS One. 2008 Jan;3(1):e1460.