
INTRODUCTION
The insulin receptor (IR) is a receptor tyrosine kinase with widespread expression that promotes glucose uptake to peripheral tissue in response to secreted insulin from the pancreatic β-cells [1]. IR signaling is also implicated in cellular growth, differentiation, survival, and metabolism [2-4], and expression of IR in β-cells is critical for the maintenance of euglycemia and islet survival [5]. In patients with type 2 diabetes mellitus, a disease characterized by hyperglycemia due to insulin insensitivity or insufficient insulin secretion, the levels of IR and associated downstream signaling pathways are perturbed [6]. In vitro experiments with the mouse insulinoma (MIN6) cell line support the importance of IR function in β-cell physiology, where knockdown of IR expression resulted in dysregulation of over 1500 genes [7]. Notable cellular functions regulated by autocrine/paracrine β-cell insulin signaling include insulin production and glucose-stimulated insulin secretion [8]. β-cell IR signaling is required in vivo for adaptive islet hyperplasia in response to pancreatic injury and insulin resistance [9], and can enhance rescue from a hyperglycemic state, as seen in diabetic rats transplanted with rat insulinoma (INS-1) cells overexpressing human IR [10]. Alternatively, adult mice with β-cell specific IR knockout (βIRKO) displayed progressive impairments in glucose-stimulated insulin secretion, glucose tolerance, and maintenance of β-cell mass [11]. Although βIRKO mice have been reported to appear phenotypically normal at birth [9, 11], fetal and newborn mice with insulin deficiency exhibited increased islet size and proliferation associated with increased pancreatic capillary density and decreased islet apoptosis [12]. Taken together, insulin/IR signaling in β-cells could be an important regulator of islet development prenatally and critical for postnatal maintenance of β-cell function. Therefore, further investigation of IR activity in β-cells is essential to better understand the pathogenesis of diabetes and areas of interest for potential therapeutics.
Pancreatic development requires temporal regulation of both transcription factor expression and external signaling pathways to generate physiologically functional adult islets [13, 14]. In particular, the structural homology between the IR and insulin-like growth factor 1 receptor (Igf-1r) enables compensation between the two receptors, allowing β-cells to utilize overlapping signaling pathways to mediate similar functions [15]. This is particularly prominent during fetal development [16]. However, the temporal importance of IR signaling on the prenatal levels of transcription factors and metabolic proteins necessary for islet, and more specifically, β-cell differentiation, proliferation, maturation, and survival has yet to be sufficiently investigated. Thus, we proposed to determine if β-cell IR is an essential regulator of β-cell development to reconcile conflicting findings from the aforementioned adult βIRKO [11] and fetal insulin [12] knockout studies, and investigate potential adaptive signaling from the homologous Igf-1r. This is the first study to investigate the temporal role of the β-cell IR on the 2nd transition period of fetal β-cell development by utilizing the mouse insulin 1 promoter to drive tamoxifen-inducible Cre-recombinase (MIP-CreER) expression in the β-cell IR knockout (MIP-βIRKO) mouse model. Our results demonstrate that β-cell specific loss of IR during fetal β-cell development results in islet overgrowth due to significantly elevated levels of Igf-2, phospho-Akt and Vegf-a signals with associated β-cell replication.
RESULTS
Characterization of MIP-βIRKO knockout mice
We investigated the temporal role of β-cell IR knockout on the 2nd transition period of fetal β-cell development (Figure 1A), a crucial stage of pancreatic development characterized by endocrine cell proliferation and fate determination. To confirm that the IR knockout was specific to pancreatic β-cells, MIP-Cre+ mice were first crossed with a B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J reporter strain. Fluorescence from the tdTomato reporter was specific to pancreatic β-cells (Figure 1B) and was not detected in the brain and other tissues, similar to results seen in other recent reports [17, 18]. The genotypes of mice were determined using PCR products of both the IR and MIP-CreER genes (Figure 1C, Supplemental Table 1). Experimental MIP-βIRKO (MIP-CreER+;IRfl/fl) mice were positive for both 313bp IRfl/fl and 268bp MIP-CreER+ (Figure 1C); heterozygous IR (IRfl/+, 313bp and 279bp) (MIP-CreER+;IRfl/+) mice were excluded. Knockout of IR in β-cells was confirmed by western blot analyses and depicted a significant reduction in IR protein level (~75% lost) in MIP-βIRKO pancreata (p < 0.05 vs. controls; Figure 1D). Reduced IR staining in insulin+ β-cells of MIP-βIRKO mouse islets was also evaluated by double immunofluorescence staining (p < 0.001 vs. control, Figure 1E).
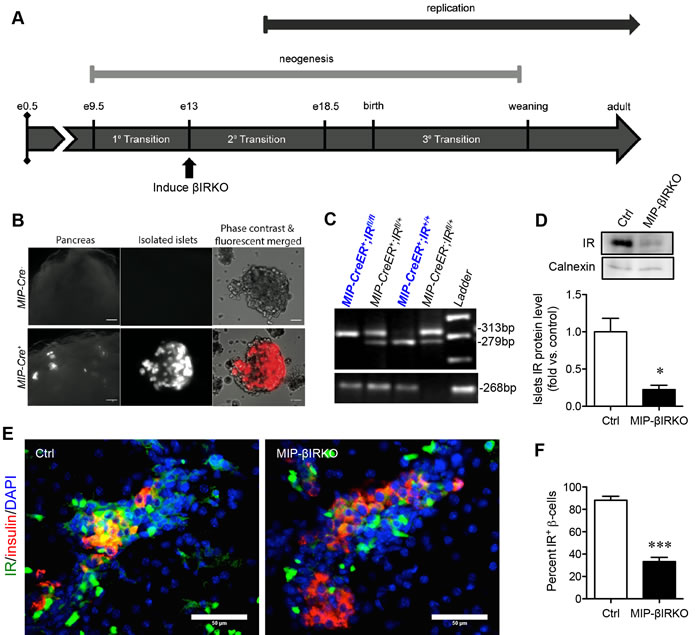
Figure 1: Confirmation of the fetal MIP-βIRKO mouse model. A. Experimental model schematic with reference timeline of important events during islet development. B. Immunofluorescence image of Cre recombinase expression within pancreatic β-cells of MIP-Cre+, but not MIP-Cre-, mouse islets following cross-breeding to a B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J reporter strain. Scale bars: 25 μm (pancreas) and 5 μm (islets). C. Genotypes of fetal mice were determined by PCR of the IR and MIP-CreER genes, followed by subsequent gel electrophoresis. D. Western blotting demonstrated a significant reduction in insulin receptor (IR) protein level of fetal MIP-βIRKO pancreata relative to controls (n = 3-4). IR protein level was normalized to calnexin and expressed as fold vs. controls. Representative blotting is shown. E. Representative double immunofluorescence images and quantification of IR+ β-cells in MIP-βIRKO relative to control pancreatic sections. Scale bar: 50 μm. White bar, control group; black bar, MIP-βIRKO group. Data are expressed as means ± SEM. *p < 0.05, ***p < 0.001 vs. controls. e = embryonic day.
Fetal MIP-βIRKO pancreas display an islet hyperplastic growth response
Body weight and blood glucose for each control and MIP-βIRKO fetus were determined at the time of dissection (e19-20), with results indicating a similar general phenotype between the two studied groups (Supplementary Figure 1). In addition, there was no significant difference in circulating plasma insulin levels or insulin content from pancreatic tissue between control and MIP-βIRKO fetal mice (Supplementary Figure 2). Immunofluorescence staining of fetal MIP-βIRKO pancreatic sections revealed a higher total number of enlarged islets per pancreatic section (Figure 2A), and morphometric analyses revealed a 1.8-fold increase in islet density (number of islets per mm2 of pancreatic sections) in fetal MIP-βIRKO pancreata (Figure 2B). The mean islet area (μm2) (p < 0.05; Figure 2C), percent islet area (percent total islet area over total pancreatic section area; p < 0.01; Figure 2D), and percent β-cell area (percent total insulin+ area over total pancreatic section area; p < 0.001; Figure 2E) were significantly increased in the fetal MIP-βIRKO pancreas when compared to control pancreata. In contrast, α-cell area (percent total glucagon+ area to total pancreatic section area) did not differ between MIP-βIRKO and control groups (Figure 2F) with no alteration of α- and β-cell distributions (Figure 2H), suggesting that the β-cell specific IR knockout does not affect α-cells and islet structure. Furthermore, no changes were noted regarding somatostatin+- and pancreatic polypeptide+-cell populations in the islets (Supplementary Figure 3).
To determine if islet changes in MIP-βIRKO pancreata were due to increased islet number or due to larger islet sizes, we tallied the size distribution of islets in both controls and MIP-βIRKO islets according to a previously established method [12]. We observed that MIP-βIRKO pancreata had a significantly decreased small islet population ( < 1000 μm2) and increased percentage of large islets ( > 10000 μm2) compared to controls (Figure 2G).
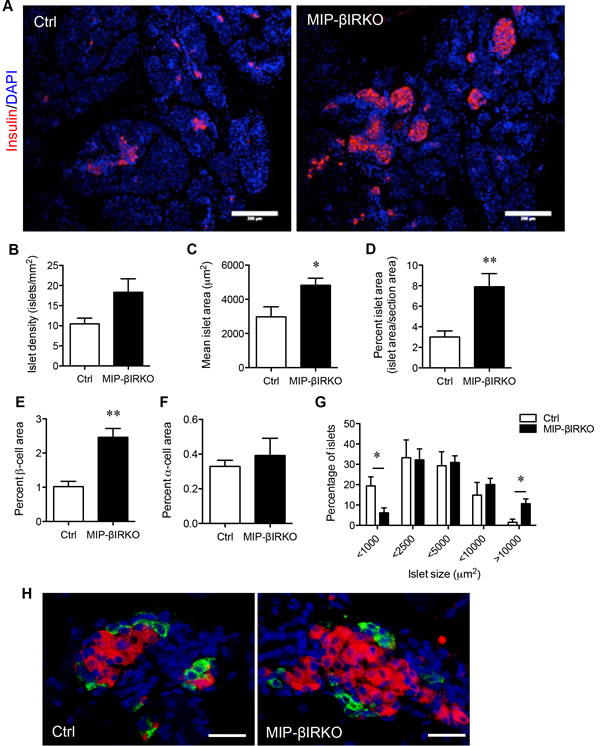
Figure 2: Fetal MIP-βIRKO mice display an islet hyperplastic growth response. A. Representative immunofluorescence image depicting the expanded population of β-cells (red) within fetal MIP-βIRKO pancreata compared to controls. Scale bar: 200 μm. Morphometric analysis of B. islet density (islets/mm2), C. mean islet area, D. percent islet area, E. percent β-cell area, and F. percent α-cell area relative to whole pancreatic section area. G. Quantification of different islet sizes. White bars, control group; black bars, MIP-βIRKO group. Data are expressed as means ± SEM (n = 5-6). *p < 0.05, **p < 0.01 vs. controls. H. Representative double immunofluorescence images for glucagon (green) and insulin (red) staining in the islets of control and MIP-βIRKO mice. Scale bar: 50 μm. Nuclei were labeled with DAPI (blue).
Fetal MIP-βIRKO increase β-cell proliferation with no change in apoptosis
To deduce a potential explanation for the islet growth response, we evaluated β-cell proliferation, death, and neogenesis in MIP-βIRKO mice. Double immunofluorescence staining for insulin and Ki67 was used to quantify the proliferating insulin+ cells. Staining showed that Ki67+/insulin+ co-localization was significantly higher (2.4-fold) in MIP-βIRKO pancreata compared to controls (p < 0.01, Figure 3A). During both fetal and postnatal development, islet neogenesis is initiated from the pancreatic ductal epithelium [19, 20]. As an indicator for islet neogenesis, we evaluated the number of ductal cells displaying early commitment to the β-cell lineage by dual labeling of Pan-CK with Pdx-1 (marker for pancreatic progenitors) or insulin (Figure 3B, 3C). The percentage of Pan-CK+ cells expressing nuclear Pdx-1 showed no significant difference between MIP-βIRKO (7%) and control (8%) pancreata (Figure 3B). A similar result was observed for the percentage of insulin+ ductal cells (Figure 3C). These findings suggest that the islet growth seen in MIP-βIRKO mice is likely attributed to the increased replication of pre-existing β-cells, rather than ductal-to-islet neogenesis.
To assess the possibility that decreased apoptosis of developing islet cells could also contribute to the islet compensatory response seen in MIP-βIRKO mice, a TUNEL assay was performed. The percentage of insulin+ cells labeled by TUNEL in MIP-βIRKO and control pancreas was similar (Figure 3D). Western blot analyses also revealed no difference in caspase-3 cleavage between MIP-βIRKO and control pancreata (Figure 3E). This data indicates that the enriched β-cell population of the MIP-βIRKO pancreas is not due to decreased β-cell apoptosis.
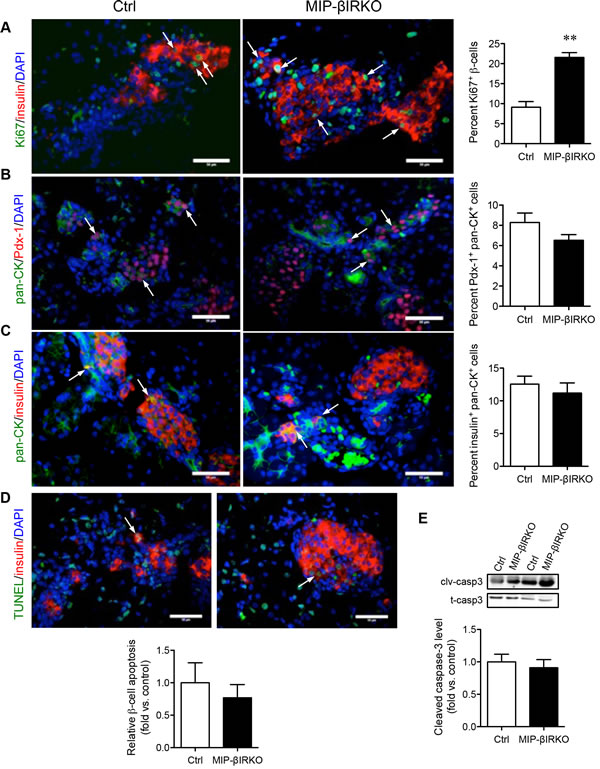
Figure 3: Fetal MIP-βIRKO mice show increased β-cell proliferation with no change in apoptosis. Representative double immunofluorescence images: A. Ki67/insulin, B. pan-CK/Pdx-1, C. pan-CK/insulin, and D. TUNEL/insulin of MIP-βIRKO and control pancreatic sections. Nuclei stain, DAPI (blue). White arrows indicate double positive cells. Scale bar: 50 μm. Quantification of Ki67+ β-cells, Pdx1+/Pan-CK+ cells, insulin+/Pan-CK+ cells and β-cell apoptosis in MIP-βIRKO islets relative to controls. E. Western blot analysis of cleaved caspase-3 (clv-casp 3) and total caspase-3 (t-casp 3). Data is normalized to T-Casp 3 and expressed as fold change to controls. White bars, control group; black bars, MIP-βIRKO group. Data are expressed as means ± SEM (n = 4-5). ***p < 0.01 vs. controls.
Fetal MIP-βIRKO islets show enhanced replication and pro-survival signaling pathway activity
We next investigated the possible mechanistic link between IR and Igf-1r signaling cascades relevant to enlarged islets in the fetal MIP-βIRKO pancreas. The PI3K/Akt signaling pathway plays a critical role in the regulation of β-cell replication [21]. We observed significantly increased phospho-Akt (S473) levels in fetal MIP-βIRKO pancreata compared to controls (p < 0.05; Figure 4A). In line with up-regulation of phospho-Akt, a significantly reduced level of phoshpo-p53 (S15), a marker for proapoptotic p53, was also observed in MIP-βIRKO pancreata relative to controls (p < 0.05; Figure 4B). To determine whether the developmental compensation for impaired β-cell IR signaling could be mediated by the action of the homologous Igf-1r signaling pathway, the levels of pancreatic Igf-1 and Igf-2 were examined. Igf-2 protein levels were significantly elevated in the pancreas of fetal MIP-βIRKO mice compared to controls (p < 0.01; Figure 4C), with an observable increase in Igf-2 staining intensity in MIP-βIRKO islets (Figure 4D). In contrast, both control and MIP-βIRKO fetal islets had low levels of Igf-1 (Figure 4E). Pancreatic immunohistochemistry also demonstrated stronger staining-intensity of Igf-1r in MIP-βIRKO islets (Figure 4F).
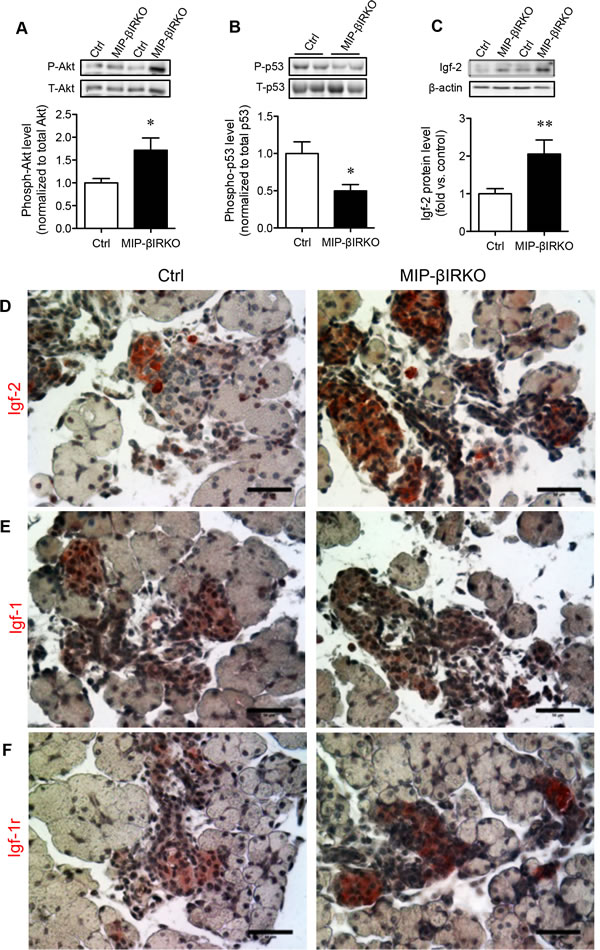
Figure 4: Fetal MIP-βIRKO islets have enhanced replication and pro-survival signaling pathway activity. Western blot analyses of phospho-S473 Akt (P-Akt) and total Akt (T-Akt) (A., n = 4), phospho-S15 p53 (P-p53) and total p53 (T-p53) (B., n = 3-4), and Igf-2 protein level (C., n = 5-13) in fetal pancreata from MIP-βIRKO and control mice. Representative blotting images are shown. White bar, control group; black bar, MIP-βIRKO group. Data are expressed as means ± SEM. *p < 0.05, **p < 0.01 vs. controls. Representative immunohistochemical images for D. Igf-2, E. Igf-1, and F. Igf-1r staining of MIP-βIRKO and control pancreatic sections. Nuclei were counterstained with hematoxylin. Scale bar: 50 μm.
Fetal MIP-βIRKO islets show increased Vegf-a levels with enriched islet-vasculature
We further evaluated whether up-regulation of Igf-2 and Akt signaling could enhance islet Vegf-a levels, a major factor associated with increased islet growth [22], and islet vascularization in the developing fetal pancreas. We found that MIP-βIRKO islets had elevated pancreatic Vegf-a protein levels as determined by immunofluorescence (Figure 5A) and western blotting (p < 0.01 vs. control islets; Figure 5B). Morphological quantification of islet vascularization (Figure 5C) was performed by measuring the PECAM-1+ area, islet capillary density, and average capillary diameter. The percentage of islet PECAM-1+ area over islet area (p < 0.01; Figure 5D) and islet capillary density (number of islet capillary per islet area) (p < 0.05; Figure 5E) were significantly increased in MIP-βIRKO mice compared to controls. No change in the average islet capillary size between MIP-βIRKO and control groups was observed (Figure 5F). In accordance with these findings, MIP-βIRKO mice demonstrated increased islet angiogenesis, as opposed to enlargement of pre-existing capillaries.
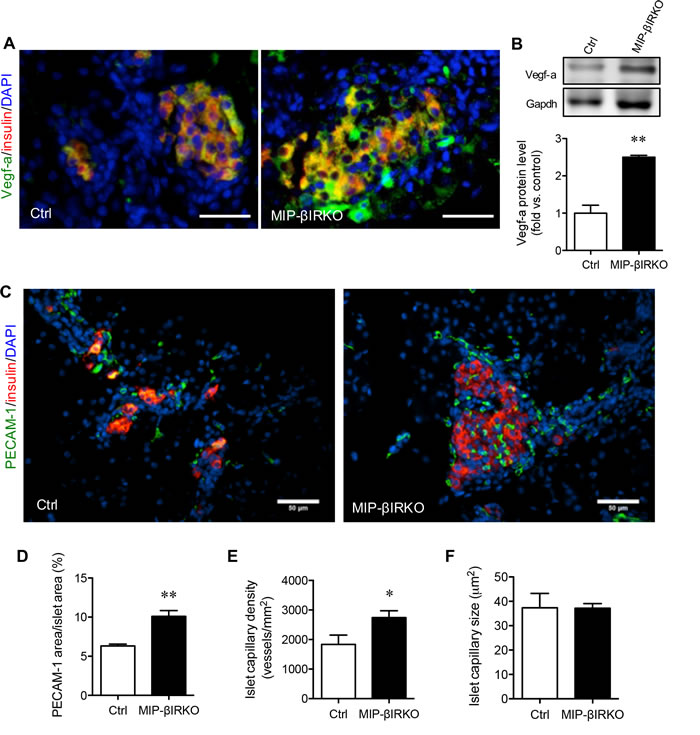
Figure 5: Fetal MIP-βIRKO islets show enhanced islet vascularization. Representative double immunofluorescence images for A. Vegf-a or C. PECAM-1 (green) with insulin (red), and nuclei stain DAPI (blue), of MIP-βIRKO and control pancreatic sections. Scale bar: 50 μm. B. Western blotting analysis of Vegf-a protein level in fetal pancreas of MIP-βIRKO and control mice (n = 3-5). Representative blotting image is shown. Quantitative analyses of D. percent PECAM-1+ area in the islets, E. islet capillary density and F. average islet capillary size in MIP-βIRKO and controls pancreata. White bars, control group; black bars, MIP-βIRKO group. Data are expressed as means ± SEM (n = 4-5). *p < 0.05, **p < 0.01 vs. controls.
MIP-βIRKO at 2nd developmental transition stage does not affect early β-cell identity
Previous studies have shown that impairments to the IR-signaling pathway result in reduced expression and nuclear-to-cytoplasmic translocation of important β-cell transcription factors, notably seen with factors Pdx-1 and MafA [7, 9]. Therefore, we employed double immunostaining on whole fetal pancreatic tissue sections to investigate potential dysregulation of transcription factors critical for development and maintenance of β-cell identity and function. Qualitative observation revealed that approximately all insulin+ cells possessed nuclear Pdx-1, Nkx6.1, Islet-1 and MafA (Figure 6) in both MIP-βIRKO and control groups. Despite the presence of larger islets in MIP-βIRKO mice, we observed similar intensity and membrane-localization of Glut-2 staining in the insulin+ cells of MIP-βIRKO and control islets (Figure 6). These results suggest that β-cell identity in MIP-βIRKO islets is maintained throughout fetal development and is independent from β-cell IR loss at the 2nd transitional stage of islet development.
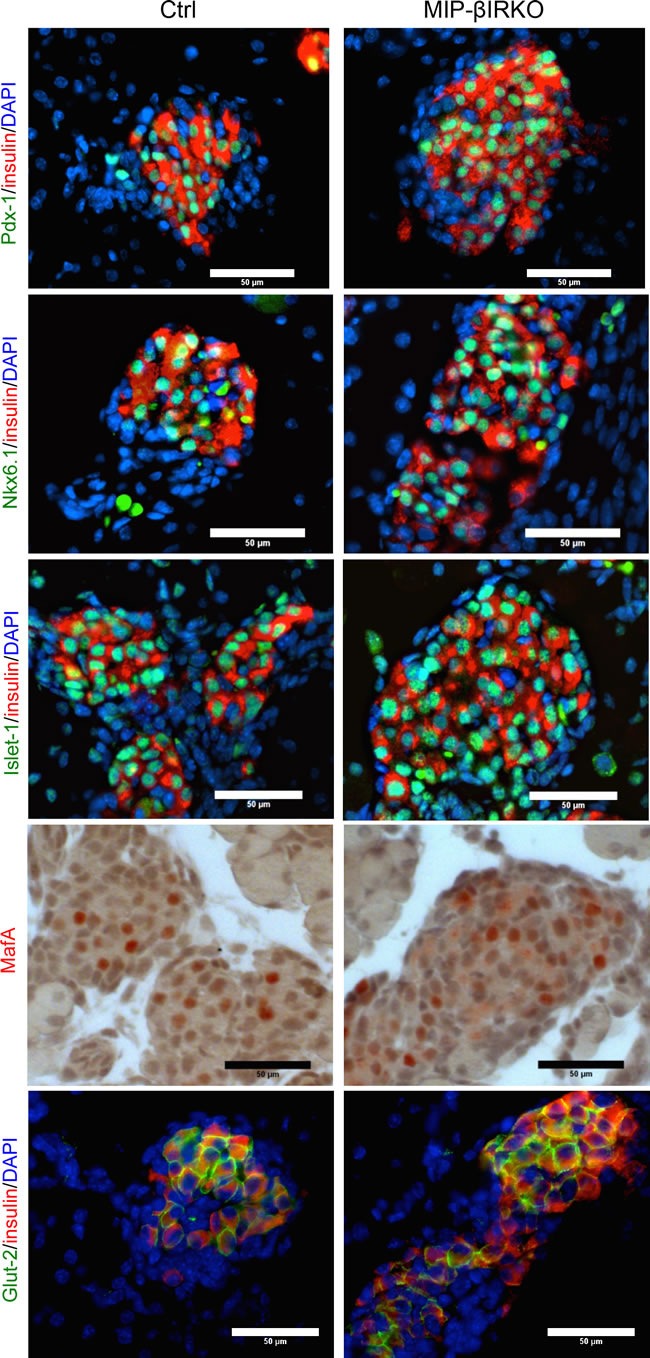
Figure 6: β-cells of fetal MIP-βIRKO islets display typical markers of β-cell identity and function. Representative double immunofluorescence images for Pdx-1, Nkx6.1, Isl-1 and Glut-2 (green) with insulin (red), and nuclei stain DAPI (blue), of MIP-βIRKO and control pancreatic sections. Immunohistochemical staining for MafA (red in nuclei). Scale bar: 50 μm.
DISCUSSION
To evaluate the in vivo role of β-cell IR activity during the 2nd transition phase of pancreatic endocrine development, we utilized a conditional and temporal MIP-βIRKO mouse model. We found that the fetal MIP-βIRKO mouse pancreas displays significantly increased mean islet and β-cell area, mainly attributed to enhanced pre-existing β-cell replication rather than islet neogenesis or apoptosis. The contribution of islet overgrowth phenomena is potentially associated with significantly enhanced Igf-2 production via Igf-1r in islets, with corresponding increased Akt phosphorylation, and enriched islet vasculature due to increased Vegf-a production (Figure 7). Our results indicate the abrupt interruption of typical autocrine/paracrine β-cell insulin signaling during the 2nd transitional phase of pancreatic endocrine development leads to an islet compensatory overgrowth.
The transcription factors Pdx-1, Nkx6.1, Isl-1, and MafA are essential for early pancreatic endocrine development, maintenance of β-cell identity, and regulation of β-cell function in mature islets, while Glut2 is necessary for glucose uptake in β-cells and regulates the appropriate insulin release response [23-26]. Similar to previous studies that examined a non-inducible βIRKO mouse model [11], we did not observe any evidence for alteration of β-cell identity in fetal MIP-βIRKO pancreata as demonstrated by insulin+ cells co-localizing to nuclear Pdx-1, Nkx6.1, Isl-1, MafA and membrane Glut2. Although previous IRKO studies suggest that the β-cell IR plays an insignificant role in fetal islet development [9, 11], the discrepancy between results from other studies and the findings presented in our own may be due to the following: (1) knockout timing during development, (2) missing in-depth characterization of islet morphology in prior reports, and (3) the previous utilization of the alternative rat insulin 2 promoter to drive Cre-recombinase expression specifically in β-cells [9, 11]. This is important because mice expressing Cre-recombinase via the rat insulin 2 promoter show ectopic Cre-recombinase expression in the brain [27], which is a key region for endocrine pancreatic regulation [28]. High-resolution microscopy analysis of a reporter line has determined that the MIP-CreER mouse model used in this study does not drive the excision of floxed genes in the central nervous system [17, 29]. It is acknowledged that ectopic human growth hormone (hGH) was highly expressed in MIP-CreER islets independent of tamoxifen administration [17], which might promote the paracrine effects of hGH-induced serotonin expression [30]. To clarify this possible influence on our results, we examined the islet morphologies of MIP-CreER negative IR knockout (MIP-CreER-;IRfl/fl) and MIP-CreER positive wide-type (MIP-CreER+;IR+/+) mouse pancreas during in utero development and did not detect any differences between these two groups. Thus, the findings indicate no major effect of hGH at the fetal development stage.
Histological analyses of islets demonstrated that fetal MIP-βIRKO β-cells have significantly increased proliferative capacity in vivo over their control β-cell counterparts. Murine β-cell neogenesis begins at e9 and remains active for the first three weeks after birth [31], and by the end of the 3rd transition stage is rapidly diminished and replaced by β-cell replication. We observed similar islet neogenesis from epithelial ducts in both MIP-βIRKO and control pancreata. Therefore, the islet compensatory response observed in MIP-βIRKO mice is likely a result of enhanced β-cell replication and expansion of individual islets as opposed to β-cell neogenesis. This is congruent with the observation of increased Akt activity, an important regulator of β-cell proliferation and anti-apoptotic signaling [21], in fetal MIP-βIRKO pancreata. The up-regulated activity of the PI3K/Akt pathway is potentially induced by elevated Igf-2 activation from compensatory Igf-1r signaling in the fetal MIP-βIRKO pancreas (Figure 7). Previous research suggests the IR and Igf-1r can compensate for one another since mice lacking both receptors specifically in β-cells died from diabetic ketoacidosis within 4-8 weeks [32]. In vitro β-cell IRKO experiments support this study’s observations with compensatory increase in Igf-1r levels [33]. Similarly, an Igf-2 compensatory response to in vivo β-cell IR loss is logical since Igf-2 levels are higher than Igf-1 during fetal development and its expression is strongly localized to islets [16]. Igf-2 is important for promoting islet cell hyperplasia and apoptosis inhibition, which would account for the changes seen in fetal MIP-βIRKO pancreata. Our findings of reduced phospho-p53 (S15) in the MIP-βIRKO pancreas, relative to controls, corroborates with our observation of upregulated active Akt in MIP-βIRKO pancreas as Akt is often correlated with decreased downstream p53 activity (Figure 7). Since active p53 is critical for cell cycle arrest and DNA repair as well as apoptosis in response to DNA damage, we speculate that impaired regulation of cell cycle arrest and subsequent DNA repair could be important for increased β-cell proliferation seen in MIP-βIRKO pancreata [34, 35]. Furthermore, this mechanism could potentially account for the β-cell atrophy that has been observed in adult βIRKO mice [11].
Our findings are consistent with studies in congenital insulin knockout mice that show increased islet proliferation and islet vascularization during fetal pancreas development [12]. MIP-βIRKO pancreata demonstrated a significant increase in the percent of PECAM-1+ area and islet capillary density, and higher Vegf-a protein levels. This data matches our observation of enhanced Igf-2 levels in MIP-βIRKO pancreata, as vascular endothelial growth factor (Vegf) can be upregulated by Igf-2 expression [36]. Igf-2 acts as a stimulatory ligand of both the Igf-1r and IR, and is therefore a critical factor for expansion of β-cell mass [37-39] and embryonic vasculogenesis by up-regulating Vegf levels via the Akt signaling pathway (Figure 7) [40]. Our recent study further determined that Vegf-a synthesis and secretion in the β-cell is regulated by the Akt/mTOR pathway, through increased HIF-1 expression, and is associated with increased β-cell mass and proliferation [22]. Since the endothelium and its associated blood supply are critical for maintenance of β-cell fate and proliferation, numerous studies have proposed that increased islet vascularization could result in enhanced β-cell replication through rapid and efficient exchange of nutrients and hormones [12, 41-44]. In fact, short-term β-cell specific Vegf-a overexpression in mice resulted in pancreatic islet hypervascularization simultaneous with increased β-cell proliferation [44]. It is important to note that Igf-2 levels rapidly diminish after birth in rodents, thus further studies are needed to examine whether the increased islet growth seen in fetal MIP-βIRKO pancreas can be sustained in postnatal life and to determine whether these mice acquire postnatal glucose intolerance or develop innate protection against age-dependent islet mass degeneration [9, 11]. We utilized multiple protocols to produce and maintain viable neonatal offspring after intraperitoneal tamoxifen administration at e13 to pregnant mothers, but all attempts were largely unsuccessful due to pup rejection or non-viable progeny. Thus, a later time-point for the temporal induction of β-cell IRKO in pregnant mothers will be required.
In summary, this is the first study to temporally knock out the β-cell IR during the 2nd transition phase of pancreatic endocrine development. Fetal MIP-βIRKO pancreata displayed an enlarged islet size and increased β-cell area that is likely associated with the observation of high β-cell proliferation in response to increased islet vascularization. Furthermore, we have demonstrated that fetal MIP-βIRKO is accompanied by heightened Igf-2/Igf-1r signaling, presumably leading to the activation of Akt and subsequent proliferation and enriched islet vasculature via increase of Vegf-a. Taken together, an understanding of the physiological function of insulin signaling on fetal β-cells is crucial for preventing abnormal islet development in utero and fine-tuning the niche for optimal in vivo survival of β-cells.
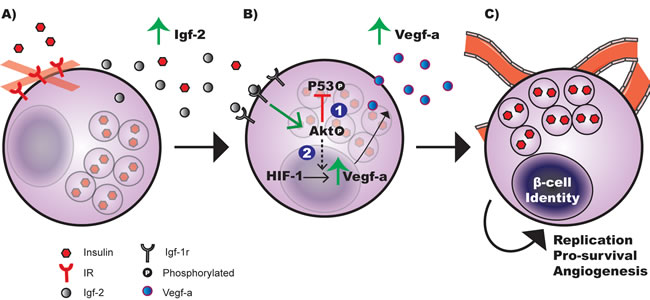
Figure 7: Working model of hyperplastic islet response observed in fetal MIP-βIRKO mice. A. The loss of insulin receptor (IR) expression on pancreatic β-cells during 2nd transition of the pancreatic development results in enhanced secretion of insulin growth factor-2 (Igf-2). B. Autocrine/paracrine activation of the insulin growth factor-1 receptor (Igf-1r) on pancreatic β-cell by Igf-2 promotes Akt phosphorylation, leading to: (1) suppressed phospho-p53 (S15) activation that prevents β-cell apoptosis, and (2) increased secretion of vascular endothelial growth factor-a (Vegf-a) through the Akt/HIF-1/Vegf-a signaling pathway in β-cells [22]. C. Together, Vegf-a-mediated hyper islet vascularization and upregulated Akt activity promotes exaggerated β-cell replication and cell survival, leading to an expanded β-cell population within the pancreas of MIP-βIRKO animals.
materials and METHODS
Generation of inducible β-cell specific insulin receptor knockout mice (MIP-βIRKO)
B6.129S4(FVB)-Insrtm1Khn/J (IRfl/fl) and Tg(Ins1-Cre/ERT)1Lphi (MIP-CreER) were obtained from the Jackson Laboratories (Bar Harbor, MA, USA; stock number: 006955) and Dr. Louis Philipson’s laboratory (University of Chicago, Chicago, IL, USA) [18], respectively. MIP-CreER and IRfl/fl mice were crossed at our facilities. The resulting MIP-CreER:IRfl/+ mice were timely mated to generate experimental fetal groups. All mice were provided ad libitum access to both food and water. All animal use protocols were approved by the Animal Use Subcommittee at Western University in accordance with the Canadian Council of Animal Care.
Tamoxifen (Sigma; St. Louis, MO, USA) was prepared by dissolving in ethanol and suspending in corn oil (Sigma). A single dose of 6mg/40g body weight was administered by i.p. injection to pregnant mice at e13 (Figure 1A). At e19-20, pregnant mice were euthanized and immediately dissected to obtain the fetal tail for genotyping and pancreas for protein extraction and morphological analyses. Isolated fetuses were measured for body weight and blood glucose, and genotype was determined using PCR primers for the Insrtm1Khn mutation and the MIP-CreER promoter (listed in Supplementary Table 1). Experimental MIP-βIRKO (MIP-CreER+;IRfl/fl) mice were MIP-CreER+ (268bp) with both IR alleles floxed by loxP sites (IRfl/fl, 313bp). Control animals included MIP-CreER- with IR alleles floxed (MIP-CreER-;IRfl/fl) and MIP-CreER+ with IR wild-type (IR+/+, 279bp) (MIP-CreER+;IR+/+). We confirmed that the phenotypes of MIP-CreER-;IRfl/fl and MIP-CreER+;IR+/+ were virtually identical in terms of islet morphology measurements.
ELISA assay for insulin
Fetal blood plasma and pancreata were collected at e19-20 from control and MIP-βIRKO mice. Blood was collected from fetal mice by decapitation and exsanguination immediately after the mother was sacrificed. Pancreatic insulin was extracted by acid-ethanol extraction buffer and homogenized. Both plasma and pancreatic insulin content were measured using an ultrasensitive (mouse) insulin ELISA kit (ALPCO, Salem, NH). Plasma insulin level was expressed as ng per ml, and pancreatic insulin content was normalized to protein and expressed as ng per mg protein [22].
Tissue processing, immunostaining, and TUNEL
Dissected fetal pancreata were fixed in 4% paraformaldehyde and embedded in paraffin as previously described [22]. Sections (2-4µm thick) were prepared and stained with appropriately diluted primary antibodies (Supplementary Table 2), and detected using fluorescently-labeled secondary antibodies (Jackson Immunoresearch Laboratories; West Grove, PA, USA). Nuclei were counterstained with DAPI (Sigma-Aldrich; St. Louis, MO, USA). Islet Igf-1, Igf-1r, Igf-2 and V-maf avian musculoaponeurotic fibrosarcoma oncogene homolog A (MafA) levels were determined by immunohistochemical staining using the streptavidin-biotin horseradish peroxidase complex and developed with aminoethyl carbazole substrate kit (Invitrogen, Burlington, ON, Canada). Apoptotic β-cells (insulin+) were identified by the terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay using an In Situ Cell Death Detection Kit (Roche Applied Science, Quebec City, QC, Canada).
Images of tissue sections were captured and islet morphologies were blindly analyzed with Image-Pro Plus software (Media Cybernetics; Rockville, MD, USA) for islet density, total islet area, islet sizes, and total α-cell or β-cell area. Islet vasculature including platelet endothelial cell adhesion molecule-1+ (PECAM-1) area per islet, islet capillary density and average islet capillary size were measured [22]. An islet was defined as a dense cell cluster containing at least 3 insulin+ cells and a glucagon+ cell [45]. Co-localization of Ki67, transcription factors, or Glut2 with insulin+ cells were also determined by double immunofluorescence or immunohistochemical staining [22].
Protein extraction and western blot analyses
Fetal pancreas, muscle, liver, and brain tissues were sonicated in Nonidet-P40 lysis buffer. Protein lysates were separated by 5-15% SDS-PAGE and were wet transferred to a nitrocellulose membrane (Bio-Rad Laboratories; Mississauga, ON, Canada), then incubated with appropriately diluted primary (Supplementary Table 2) and secondary horseradish peroxidase-conjugated secondary antibodies. Proteins were visualized with Western Lightning Plus-Enhanced Chemoluminescence reagents (PerkinElmer; Waltham, MA, USA). All images were recorded using a Versadoc Imaging System (Bio-Rad Laboratories). Densitometric analyses of images were performed at subsaturation levels with Image Lab 3.0 software (Bio-Rad Laboratories) and normalized to appropriate loading controls.
Statistical analyses
Statistical analyses were performed using either one-way ANOVA and Bonferonni’s multiple comparison tests or Student’s unpaired t-test with GraphPad Prism 6 (GraphPad Software; La Jolla, CA, USA). Data is presented as means ± SEM. Differences in results were considered statistically significant when p < 0.05.
ACKNOWLEDGMENTS
The authors sincerely thank Dr. Louis Philipson from University of Chicago, Chicago, IL, USA for providing MIP-CreER mouse model; and Mr. Jinming Li for his technical assistance and support.
CONFLICTS OF INTERESTS
The authors have no conflicts to disclose.
GRANT SUPPORT
This work was supported by grants from the Canadian Institutes of Health Research (CIHR, Grant No. MOP 89800). LZ is a recipient of a Canadian Institutes of Health Research Master Student Research Award.
REFERENCES
1. Belfiore A, Frasca F, Pandini G, Sciacca L, and Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009; 30: 586-623. doi: 10.1210/er.2008-0047
2. Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH 3rd, Wright CV, White MF, Arden KC, and Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic β cell growth. J Clin Invest. 2002; 110: 1839-47.
3. Scioli MG, Cervelli V, Arcuri G, Gentile P, Doldo E, Bielli A, Bonanno E, and Orlandi A. High insulin-induced down-regulation of Erk-1/IGF-1R/FGFR-1 signaling is required for oxidative stress-mediated apoptosis of adipose-derived stem cells. J Cell Physiol. 2014; 229: 2077-87. doi: 10.1002/jcp.24667
4. Liew CW, Assmann A, Templin AT, Raum JC, Lipson KL, Rajan S, Qiang G, Hu J, Kawamori D, Lindberg I, Philipson LH, Sonenberg N, Goldfine AB, et al. Insulin regulates carboxypeptidase E by modulating translation initiation scaffolding protein eIF4G1 in pancreatic cells. Proc Natl Acad Sci U S A. 2014; 111: E2319-28. doi: 10.1073/pnas.1323066111
5. Johnson JD, Bernal-Mizrachi E, Alejandro EU, Han Z, Kalynyak TB, Li H, Beith JL, Gross J, Warnock GL, Townsend RR, Permutt MA, and Polonsky KS. Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci U S A. 2006; 103:19575-80.
6. Folli F, Okada T, Perego C, Gunton J, Liew CW, Akiyama M, D’Amico A, La Rosa S, Placidi C, Lupi R, Marchetti P, Sesti G, Hellerstein M, et al. Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS One. 2011; 6: e28050. doi: 10.1371/journal.pone.0028050
7. Ohsugi M, Cras-Méneur C, Zhou Y, Bernal-Mizrachi E, Johnson JD, Luciani DS, Polonsky KS, and Permutt MA. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J Biol Chem. 2005; 280: 4992-5003.
8. Xu GG, Gao ZY, Borge PD Jr, Jegier PA, Young RA, amd Wolf BA. Insulin expression, Ca(2+) homeostasis, and insulin expression and secretion. Biochemistry. 2000; 39: 14912-9.
9. Okada T, Liew CW, Hu J, Hinault C, Michael MD, Krtzfeldt J, Yin C, Holzenberger M, Stoffel M, and Kulkarni RN. Insulin receptors in β-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci U S A. 2007; 104: 8977-82.
10. Kim MH, Hong SH, and Lee MK. Insulin receptor-overexpressing β-cells ameliorate hyperglycemia in diabetic rats through Wnt signaling activation. PLoS One. 2013; 8: e67802. doi:10.1371/journal.pone.0067802
11. Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, and Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999; 96: 329-39.
12. Duvillié B, Currie C, Chrones T, Bucchini D, Jami J, Joshi RL, and Hill DJ. Increased islet cell proliferation, decreased apoptosis, and greater vascularization leading to β-cell hyperplasia in mutant mice lacking insulin. Endocrinology. 2002; 143: 1530-37.
13. Habener JF, Kemp DM, and Thomas MK. Minireview: transcriptional regulation in pancreatic development. Endocrinology. 2005; 146: 1025-34.
14. Gittes GK. Developmental biology of the pancreas: a comprehensive review. Dev Biol. 2009; 326: 4-35. doi: 10.1016/j.ydbio.2008.10.024
15. Nakae J, Kido Y, and Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001; 22: 818-835.
16. Kulkarni RN. New insights into the roles of insulin/IGF-I in the development and maintenance of β-cell mass. Rev Endocr Metab Disord. 2005; 6: 199-210.
17. Oropeza D, Jouvet N, Budry L, Campbell JE, Bouyakdan K, Lacombe J, Perron G, Bergeron V, Neuman JC, Brar HK, Fenske RJ, Meunier C, Sczelecki S, et al. Phenotypic characterization of MIP-CreERT1Lphi mice with transgene-driven islet expression of human growth hormone. Diabetes. 2015; 64: 3798-807. doi: 10.2337/db15-0272
18. Tamarina NA1, Roe MW, Philipson L. Characterization of mice expressing Ins1 gene promoter driven CreERT recombinase for conditional gene deletion in pancreatic β-cells. Islets. 2014; 6: e27685. doi: 10.4161/isl.27685
19. Dodge R, Loomans C, Sharma A, and Bonner-Weir S. Developmental pathways during in vitro progression of human islet neogenesis. Differentiation. 2009; 77: 135-47. doi: 10.1016/j.diff.2008.09.015
20. Al-Hasani K, Pfeifer A, Courtney M, Ben-Othman N, Gjernes E, Vieira A, Druelle N, Avolio F, Ravassard P, Leuckx G, Lacas-Gervais S, Ambrosetti D, Benizri E, Hecksher-Sorensen J, Gounon P, Ferrer J, Gradwohl G, Heimberg H, Mansouri A, and Collombat P. Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell. 2013; 26: 86-100. doi: 10.1016/j.devcel.2013.05.018
21. Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, Polonsky KS, Naji A, and Birnbaum MJ. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001; 7: 1133-7.
22. Feng ZC, Popell A, Li J, Silverstein J, Oakie A, Yee SP, and Wang R. c-Kit Receptor Signaling Regulates Islet Vasculature, β-Cell Survival, and Function In Vivo. Diabetes. 2015. 64: 3852-3866.
23. Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Cruz Dela F, Schwitzgebel V, Hayes-Jordan A, and German M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of β-cell formation in the pancreas. Development. 2000; 127: 5533-40.
24. Ediger BN, Du A, Liu J, Hunter CS, Walp ER, Schug J, Kaestner KH, Stein R, Stoffers DA, and May CL. Islet-1 is essential for β-cell function. Diabetes. 2014; 63: 4206-17. doi: 10.2337/db14-0096
25. Seufert J, Weir GC, and Habener JF. Differential expression of the insulin gene transcriptional repressor CCAAT/enhancer-binding protein β and transactivator islet duodenum homeobox-1 in rat pancreatic β cells during the development of diabetes mellitus. J Clin Invest. 1998; 101: 2528-39.
26. Nishimura W, Takahashi S, and Yasuda K. MafA is critical for maintenance of the mature β cell phenotype in mice. Diabetologia. 2014; 58: 566-74. doi: 10.1007/s00125-014-3464-9
27. Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, Bernal-Mizrachi E, Elghazi L, Roe MW, et al. Conditional gene targeting in mouse pancreatic β-cells: analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010; 59: 3090-8. doi: 10.2337/db10-0624
28. Schwartz MW, Seeley RJ, Tschöp MH, Woods SC, Morton GJ, Myers MG, and D’Alessio D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature. 2013; 503: 59-66. doi: 10.1038/nature12709
29. Cheng Y, Su Y, Shan A, Jiang X, Ma Q, Wang W, Ning G, and Cao Y. Generation and characterization of transgenic mice expressing mouse Ins1 promoter for pancreatic β-cell-specific gene overexpression and knockout. Endocrinology. 2015;156: 2724-31. doi: 10.1210/en.2015-1104
30. Brouwers B, de Faudeur G, Osipovich AB, Goyvaerts L, Lemaire K, Boesmans L, Cauwelier EJ, Granvik M, Pruniau VP, Van Lommel L, Van Schoors J, Stancill JS, Smolders I, et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab. 2014; 20: 979-90. doi: 10.1016/j.cmet.2014.11.004
31. Gunasekaran U, Hudgens CW, Wright BT, Maulis MF, and Gannon M. Differential regulation of embryonic and adult β cell replication. Cell Cycle. 2012; 11: 2431-42. doi: 10.4161/cc.20545
32. Xuan S, Szabolcs M, Cinti F, Perincheri S, Accili D, and Efstratiadis A. Genetic analysis of type-1 insulin-like growth factor receptor signaling through insulin receptor substrate-1 and -2 in pancreatic β cells. J Biol Chem. 2010; 285: 41044-50. doi: 10.1074/jbc.M110
33. Assmann A, Ueki K, Winnay JN, Kadowaki T, and Kulkarni RN. Glucose effects on β cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol Cell Biol. 2009; 29: 3219-28. doi: 10.1128/MCB.01489-08
34. Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, Iwai-Kanai E, Ikeda K, Ueyama T, Ogata T, and Matoba S. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc Natl Acad Sci U S A. 2014; 111: 3116-21. doi: 10.1073/pnas.1318951111
35. Wrede CE, Dickson LM, Lingohr MK, Briaud I, and Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic β-cells (INS-1). J Biol Chem. 2002; 277: 49676-84.
36. Lee SR, Kim SH, Chae HD, Kim CH, and Kang BM. Influence of vascular endothelial growth factor on the expression of insulin-like growth factor-II, insulin-like growth factor binding protein-2 and 5 in human luteinized granulosa cells. Gynecol Endocrinol. 2012; 28: 917-20. doi: 10.3109/09513590.2012.683070
37. Modi H, Cornu M, and Thorens B. Glutamine stimulates biosynthesis and secretion of insulin-like growth factor 2 (IGF2), an autocrine regulator of β-cell mass and function. J Biol Chem. 2014; 289: 31972-82. doi: 10.1074/jbc.M114.587733
38. Estil les E, Téllez N, Escoriza J, and Montanya E. Increased β-cell replication and β-cell mass regeneration in syngeneically transplanted rat islets overexpressing insulin-like growth factor II. Cell Transplant. 2012; 21: 2119-29. doi: 10.3727/096368912X638955
39. Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, and Accili D. Role of the forkhead protein FoxO1 in β cell compensation to insulin resistance. J Clin Invest. 2006; 116: 775-82.
40. Piecewicz SM, Pandey A, Roy B, Xiang SH, Zetter BR, and Sengupta S. Insulin-like growth factors promote vasculogenesis in embryonic stem cells. PLoS One. 2012; 7: e32191. doi:10.1371/journal.pone.0032191
41. Reinert RB, Brissova M, Shostak A, Pan FC, Poffenberger G, Cai Q, Hundemer GL, Kantz J, Thompson CS, Dai C, McGuinness OP, and Powers AC. Vascular endothelial growth factor-a and islet vascularization are necessary in developing, but not adult, pancreatic islets. Diabetes. 2013; 62: 4154-64. doi: 10.2337/db13-0071
42. Takenouchi K, Shrestha B, Yamakuchi M, Yoshinaga N, Arimura N, Kawaguchi H, Nagasato T, Feil R, Kawahara K, Sakamoto T, Maruyama I, and Hashiguchi T. Upregulation of non-β cell-derived vascular endothelial growth factor A increases small clusters of insulin-producing cells in the pancreas. Exp Clin Endocrinol Diabetes. 2014; 122: 308-15. doi: 10.1055/s-0034-1371811
43. Guo T, Landsman L, Li N, and Hebrok M. Factors expressed by murine embryonic pancreatic mesenchyme enhance generation of insulin-producing cells from hESCs. Diabetes. 2013; 62: 1581-92. doi: 10.2337/db12-0167
44. De Leu N, Heremans Y, Coppens V, Van Gassen N, Cai Y, D’Hoker J, Magenheim J, Salpeter S, Swisa A, Khalaileh A, Arnold C, Gradwohl G, Van de Casteele M, et al. Short-term overexpression of VEGF-A in mouse β cells indirectly stimulates their proliferation and protects against diabetes. Diabetologia. 2014; 57: 140-47.
45. Wang RN, Bouwens L, and Klöppel G. Beta-cell proliferation in normal and streptozotocin-treated newborn rats: site, dynamics and capacity. Diabetologia. 1994; 37: 1088-96.