
INTRODUCTION
Activation of the epidermal growth factor receptor (EGFR) signaling pathway is known to play a significant role in the pathophysiology of non-small cell lung cancer (NSCLC)[1]. It may also be important in other tumors[2]. Aberrant activation of the signaling pathway may occur due to mutations in exons 18 through 21, which encode part of the tyrosine kinase domain and are bundled around the ATP-binding pocket of the enzyme[1-4]. There is a broad literature on the efficacy of EGFR inhibitors in NSCLC[3-6] . Although many patients with EGFR-mutant NSCLC respond to EGFR inhibitors initially, they eventually develop resistance to treatment. Therefore, combination approaches to overcome resistance is an area of active clinical research[7, 8].
The role of EGFR inhibition in patients with wild-type EGFR and lung cancer has been debated. Studies with erlotinib show increased survival in unselected patients with lung cancer,[9] though there is a general consensus that patients with sensitive EGFR mutations are most likely to benefit[3, 4]. Recently, preclinical studies have demonstrated that EGFR can signal via a kinase-independent pathway[10], suggesting a role for combining EGFR kinase inhibitors and antibodies. Furthermore, preclinical models suggest that several molecules synergize with EGFR inhibitors, including the multikinase inhibitor dasatinib[11] and the proteasome inhibitor bortezomib[12]. Herein, we report our experience with EGFR-based combination regimens in patients with advanced, heavily-pretreated NSCLC referred to a phase I clinic, including those with secondary resistance to erlotinib, resistant mutations, and EGFR wild-type disease.
RESULTS
EGFR mutations
Twenty-one of 131 NSCLC patients (16%) tested had EGFR mutations. Twenty-five EGFR mutations were present in those 21 individuals. Four patients had two EGFR mutations. Ten of the 25 EGFR mutations were present in exon 19; three in exon 20; and, 12 in exon 21. Of the four patients who had two EGFR mutations, three of them had two EGFR mutations in exon 21 and 1 patient had an EGFR mutation in exon 19 and exon 20. Deletions in exon 19 (n = 9) and the L858R substitution mutation in exon 21 (n = 7) were the two most common types of mutations.
Table 1: Baseline characteristics of 15 evaluable patients with EGFR mutation-positive NSCLC and 24 patients with EGFR wild-type NSCLC treated with EGFR inhibitor-based combination regimens
Variable |
EGFR-mutant n=15 |
EGFR wild-type n=24 |
Sex, n (%) |
||
Male |
8 (53) |
14 (58) |
Female |
7 (47) |
10 (42) |
Age (years) |
||
Median |
65 |
69 |
Range |
29-76 |
42-82 |
Ethnicity, n (%) |
||
Caucasian |
6 (40) |
21 (88) |
Asian |
5 (33) |
0 (0) |
Hispanic |
2 (13) |
2 (8) |
African American |
2 (13) |
1 (4) |
Histology, n (%) |
||
Adenocarcinoma |
13 (87) |
20 (83) |
Squamous cell |
1 (7) |
3 (13) |
Adenosquamous |
1 (7) |
0 (0) |
Neuroendocrine |
0 (0) |
1 (4) |
EGFR mutation, n (%) |
||
Exon 19 |
6 (40) |
0 (0) |
Exon 20 |
2 (13) |
0 (0) |
Exon 21 |
4 (27) |
0 (0) |
Two mutations |
3 (20) |
0 (0) |
KRAS mutation, n (%) |
||
Positive |
0 (0) |
2 (8) |
Negative |
13 (87) |
18 (75) |
Unknown |
2 (13) |
4 (17) |
PIK3CA mutation, n (%) |
||
Positive |
2 (13) |
2 (8) |
Negative |
5 (33) |
11 (46) |
Unknown |
8 (53) |
11 (46) |
History of smoking, n (%) |
||
Ex-smoker |
7 (47) |
16 (67) |
Never smoked |
8 (53) |
8 (33) |
Number of prior therapies |
||
Median |
4 |
2 |
Range |
0-7 |
1-7 |
Previous EGFR inhibitors, n (%) |
||
Yes |
12 (80) |
8 (33) |
No |
3 (20) |
16 (67) |
ECOG PS |
||
0 |
4 (27) |
5 (21) |
1 |
10 (67) |
14 (58) |
2 |
1 (7) |
5 (21) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; EGFR, Epidermal Growth Factor Receptor; KRAS, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; NSCLC, Non-Small Cell Lung Cancer; PS, Performance status; PIK3CA, Phosphatidylinositol 3-kinase, catalytic, alpha polypeptide; PR, Partial response; SD, Stable disease
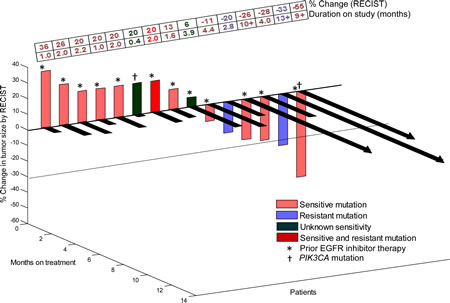
Figure 1: 3-D Waterfall plot. Best response by RECIST, of 15 NSCLC patients with EGFR positive-mutations treated with an EGFR inhibitor-based regimen. Patients with clinical progression or with new metastases were graphed as 20% progression. Time to treatment failure in months is represented by solid lines and the arrow indicates that the patient was still on study when the data was censored. Patients with PIK3CA mutations in addition to EGFR mutation and patients who received prior EGFR inhibitor therapy are designated as such. Dotted horizontal line at -30% indicates border for partial response.
Treatment
Fifteen of the 21 patients (71%) with an underlying EGFR mutation were enrolled in five clinical trials that included an EGFR inhibitor combination (Patients and Methods and Table 2).
Of the remaining six EGFR-mutant NSCLC patients, one patient was treated on a clinical trial that did not include an EGFR inhibitor, one patient was referred to hospice, one patient died soon after being seen, and three patients were treated with single-agent erlotinib by their primary oncologist or on study.
Patients treated with EGFR inhibitor-based combinations
Patient characteristics of the 15 EGFR mutation-positive and 24 EGFR wild-type NSCLC patients treated with EGFR inhibitor-based combination regimens are summarized in Table 1.
Co-existing mutations in 15 EGFR-mutant patients treated with EGFR-based regimens
Simultaneous mutations in other genes were assessed when tissue was available. Two of seven EGFR mutation-positive patients (29%) assessed had a PIK3CA mutation. One patient (case #15, Table 2) had an E545K mutation in exon 9 of the PIK3CA gene in addition to the EGFR mutation (T847I in exon 21; unknown sensitivity to EGFR inhibitors). A second patient (case #5, Table 2) had an E542K mutation in exon 9 of the PIK3CA gene in addition to two known sensitive EGFR mutations (L858R and G873E) in exon 21. No patient that underwent treatment with an EGFR inhibitor-based combination had a KRAS mutation (though one patient who was not treated had a G12C mutation in addition to a resistant EGFR [D761N] mutation in exon 19).
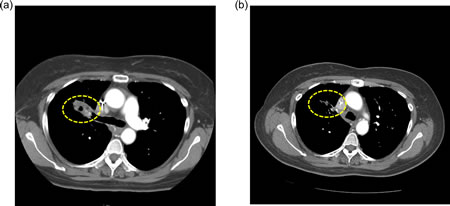
Figure 2: Computed tomography (CT) scans of a NSCLC patient (case #5, Table 2) with two sensitive EGFR mutations (L858R in exon 21 and G873E in exon 21) and a PIK3CA mutation (E542K in exon 9). a) CT at baseline, and b) CT taken 5 months after treatment initiation with erlotinib/cetuximab/bevacizumab demonstrating a PR (-55%). Duration of response = 9+ months. Patient had received prior erlotinib for 14.3 months.
Other mutations in EGFR wild-type patients treated with EGFR-based regimens
Two of 13 patients (15%) with EGFR wild-type disease assessed for PIK3CA mutation had an E545K mutation in exon 9 of the PIK3CA gene (cases #15 and 23, Table 3). Two of 20 patients (10%) with EGFR wild-type evaluated for KRAS mutation had a G12D mutation (cases #20 and 21, Table 3). Of the two patients with EGFR wild-type disease evaluated for TP53 mutation, one had an R196 mutation in exon 6 (case #1, Table 3) and the other had a V157F mutation in exon 5 (case #19, Table 3).
Responses to EGFR inhibitor-based combinations in EGFR-mutant NSCLC patients
Three of 15 evaluable (20%) patients attained either PR (n=2; cases #2 and 5, Table 2) or SD ≥6 months (n=1; case #10, Table 2). Six patients came off study prior to post-treatment imaging evaluation due to clinical progression (all of whom were arbitrarily graphed as 20% progression in Figure 1). One patient (case #2, Table 2) achieved a PR (33% decrease; duration=13+ months) on erlotinib/cetuximab despite having a known EGFR-resistant mutation (insertion in exon 20)[13]. This patient had previously received two lines of standard chemotherapy but had not received prior EGFR inhibitor therapy. TTF on the last standard treatment before referral to phase I was 2.0 months. A second patient (case #5, Table 2) with two known EGFR-sensitive mutations (L858R and G873E in exon 21) and a PIK3CA mutation (E542K in exon 9) had a PR (55% decrease; duration=9+ months) on erlotinib/cetuximab/bevacizumab. This patient had received six lines of prior therapy including single-agent erlotinib (TTF=14.3 months). TTF on the last standard treatment before referral was 4.5 months. A third patient (case #10, Table 2) with a known EGFR-sensitive mutation (L858R) in exon 21 attained SD for 10+ months on erlotinib/bortezomib. This patient had received six lines of prior therapy including single-agent erlotinib (TTF = 5.5 months). TTF on the last standard treatment before referral was 5.9 months.
Responses and TTF in EGFR-mutant patients who had received prior EGFR inhibitors
Of the 12 patients who had progressed previously on EGFR inhibitors and received an EGFR inhibitor-based combination regimen after referral, two patients (17%) achieved either PR (n=1; case #5, Table 2; duration=9+ months) or SD≥6 months (n=1; case #10, Table 2; duration=10+ months) on this study. Of the three patients who achieved SD ≥6 months/PR on this study, two patients (67%; cases #10 and #5) had received prior erlotinib therapy as single-agent and had progressed. The TTF on the EGFR inhibitor-based combination therapies on this study is ongoing at 10+ and 9+ months vs. 5.5 and 14.3 months respectively on prior single-agent erlotinib. For the 12 EGFR-mutant patients who received prior EGFR inhibitors, median TTF on their EGFR inhibitor-based combination regimen after referral was 2 months as compared to 8 months on a prior EGFR inhibitor (p=0.044)
Responses to EGFR inhibitor-based combinations in NSCLC patients with EGFR wild-type disease
Of the 24 patients with EGFR wild-type disease (Table 3) treated on the same protocols as listed above, 19 were evaluable for response to treatment. Five patients were not evaluable for response, as restaging had not yet occurred at the time of this analysis. Overall, 5 of 19 evaluable patients (26%) had either a PR (n=1, case #12, Table 3; duration = 4.1 months) or SD ≥6 months (n=4; cases #1, 6, 9, and, 10; duration = 26.5 9.2, 6.5, and 11.0 months respectively). All of them were treated on erlotinib/cetuximab/bevacizumab. The median TTF of the 24 patients with EGFR wild-type disease treated on EGFR inhibitor-based combinations in phase I clinical trial program is statistically longer (3.3 months; 95% CI, 1.2-5.4 months) compared to median TTF on last standard therapy (2.3 months; 95% CI, 1.8-2.8 months; p=0.045; Figure 3). Four of five patients with SD ≥6 months/PR had not received prior EGFR inhibitors. One patient (case #6, Table 3), who had previously received erlotinib as a single-agent for 6.2 months and had progressed, had SD for 9.2 months on erlotinib/cetuximab/bevacizumab. One of five patients who achieved SD ≥6 months/PR had squamous cell histology (case #1, Table 3; duration of SD=26.5 months). Overall, two evaluable patients with EGFR wild-type disease (cases #1 and 14, Table 3) who were treated with EGFR inhibitors had squamous histology.
Responses in NSCLC patients with squamous cell histology
A total of three evaluable patients treated on EGFR inhibitor-based combinations had squamous cell histology, two of whom had EGFR wild-type disease (cases #1 and 14, Table 3), and one of whom had an EGFR mutation (case #15, Table 2). One of the two patients with wild-type EGFR (case #1, Table 3) attained SD for 26.5 months on erlotinib/cetuximab/bevacizumab. This patient had received two lines of standard therapy, but was not previously treated with an EGFR inhibitor. TTF on the last standard therapy prior to this study was 2.4 months. A second patient with wild-type EGFR (case #14, Table 3) had progressive disease after 2.1 months on erlotinib/cetuximab/bevacizumab. This patient had received only one line of standard therapy (not an EGFR inhibitor) and the TTF was 4.2 months. The third patient was EGFR mutation-positive (case #15, Table 2; T847I, unknown sensitivity to EGFR inhibitors) and also had an additional PIK3CA mutation (E545K in exon 9); this patient had progressive disease within one month on erlotinib and dasatinib. This patient had received two lines of standard therapy, but not an EGFR inhibitor. TTF on last therapy before referral was 1.0 month.
Table 2: Characteristics of 15 patients with EGFR mutations§ treated with EGFR inhibitor-based regimens
Case No. |
Histology |
EGFR mutations (exon) |
Sensitive/ Resistant† |
Concomitant mutations |
Previous EGFR inhibitor therapy |
Treatment in phase I program |
||||
Yes/ No |
Best response |
TTF (months) |
EGFR inhibitor |
Best response |
TTF (months) |
|||||
1 |
Adenocarcinoma |
insertion/ deletion in exon 19 |
Sensitive |
PIK3CA: Not done* TP53: Not done |
Yes |
PR |
24.8 |
erlotinib, dasatinib |
PD** |
2 |
2 |
Adenocarcinoma |
insertion in exon 20 |
Resistant |
PIK3CA: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab |
PR |
13+ |
3 |
Adenocarcinoma |
deletion in exon 19 |
Sensitive |
PIK3CA:Not done TP53: Not done |
Yes |
SD |
12.0 |
erlotinib, cetuximab, bevacizumab |
SD |
4 |
4 |
Adenocarcinoma |
deletion in exon 19 |
Sensitive |
PIK3CA:Not done TP53: Not done |
Yes |
PR |
26.1 |
erlotinib, cetuximab |
PD** |
1 |
5 |
Adenocarcinoma |
L858R (exon 21), G873E (exon 21) |
Sensitive, Sensitive |
PIK3CA: E542K (exon 9) TP53: Not done* |
Yes |
SD |
14.3 |
erlotinib, cetuximab, bevacizumab |
PR |
9+ |
6 |
Adenocarcinoma |
D830Y (exon 21), V834A (exon 21) |
Unknown significance |
PIK3CA:No TP53: Not done |
Yes |
SD |
4.0 |
cetuximab, sirolimus |
SD |
4 |
7 |
Adenocarcinoma |
L858R (exon 21) |
Sensitive |
PIK3CA:No TP53: Not done |
Yes |
PR |
8.1 |
erlotinib, bortezomib |
SD |
2 |
8 |
Adenocarcinoma |
T790M (exon 20), deletion in exon 19 |
Resistant, Sensitive |
PIK3CA: Not done* TP53: Not done |
Yes |
PR |
1.9 |
erlotinib, cetuximab |
PD** |
2 |
9 |
Adenocarcinoma |
L858R (exon 21) |
Sensitive |
PIK3CA:No TP53: Not done |
Yes |
SD |
11.5 |
erlotinib, dasatinib |
PD |
2 |
10 |
Adenocarcinoma |
L858R (exon 21) |
Sensitive |
PIK3CA:Not done TP53: Not done |
Yes |
PR |
5.5 |
erlotinib, bortezomib |
SD |
10+ |
11 |
Adenocarcinoma |
insertion in exon 20 |
Resistant |
PIK3CA: Not done* KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
SD |
3 |
12 |
Adenocarcinoma |
deletion in exon 19 |
Sensitive |
PIK3CA:No TP53: Not done |
Yes |
SD |
3.5 |
cetuximab, sirolimus |
SD |
4 |
13 |
Adenosquamous |
deletion in exon 19 |
Sensitive |
PIK3CA: Not done* TP53: Not done* |
Yes |
PR |
3.4 |
erlotinib, cetuximab |
PD |
1 |
14 |
Adenocarcinoma |
deletion in exon 19 |
Sensitive |
PIK3CA:Not done TP53: Not done |
Yes |
SD |
8.0 |
erlotinib, cetuximab |
PD** |
2 |
15 |
Squamous cell carcinoma |
T847I (exon 21) |
Unknown significance |
PIK3CA: E545K (exon 9) TP53: Not done* |
No |
NA |
NA |
erlotinib, dasatinib |
PD** |
0.4 |
Abbreviations: EGFR, Epidermal Growth Factor Receptor; KRAS, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; NA, Not applicable; NSCLC, Non-Small Cell Lung Cancer; PR, Partial response; PIK3CA, Phosphatidylinositol 3-kinase, catalytic, alpha polypeptide; PD, Progressive disease; SD, Stable disease; TTF, Time to treatment failure; TP 53, Tumor Protein p53
§ Of the remaining six EGFR-mutant NSCLC patients who were not treated on EGFR inhibitor-based regimens, two patients had a sensitive deletion in exon 19, one patient had a resistant D761N mutation in exon 19, one patient had 2 EGFR-sensitive mutations, L858R and L833V, in exon 21, and two patients had a sensitive L858R mutation in exon 21
†Sensitive or resistant is denoted based on survey of the literature.
*not done because tissue was not available for molecular analysis
**clinical progression/new metastasis
+did not progress at the time of analysis
Responses in NSCLC patients with other simultaneous mutations
PIK3CA:
Of the two EGFR mutation-positive patients with a simultaneous PIK3CA mutation, one patient (case #5, Table 2) with an E542K mutation in exon 9 of the PIK3CA gene in addition to two sensitive EGFR mutations (L858R and G873E in exon 21), achieved a PR (-55%) for 9+ months on erlotinib/cetuximab/bevacizumab. The other EGFR mutation-positive patient (T847I in exon 21; unknown sensitivity to EGFR inhibitors) with a co-existing E545K mutation in exon 9 of the PIK3CA gene (case #15, Table 2), had progressive disease in 0.4 months on erlotinib and dasatinib.
Of the two EGFR wild-type NSCLC patients with a E545K mutation in exon 9 of the PIK3CA gene, one patient (case #15, Table 3) has ongoing SD at 3+ months on erlotinib/cetuximab/bevacizumab; the other patient (case #23, Table 3) had SD for 3 months on erlotinib/dasatinib.
Table 3: Characteristics of 24 NSCLC patients with EGFR wild-type disease treated with EGFR inhibitor-based regimens
Case Number |
Histology |
Other mutations |
Previous EGFR inhibitor therapy |
Treatment in phase I program |
||||
Yes/No |
Best response |
TTF (months) |
EGFR inhibitor-based therapy |
Best response |
TTF (months) |
|||
1 |
Squamous cell carcinoma |
PIK3CA: No KRAS: No TP53: R196 (exon 6) |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
SD |
26.5 |
2 |
Adenocarcinoma |
PIK3CA: Not done KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
PD** |
2.0 |
3 |
Adenocarcinoma |
PIK3CA: Not done KRAS: Not done TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
PD** |
0.9 |
4 |
Adenocarcinoma |
PIK3CA: Not done* KRAS: No TP53: Not done |
Yes |
PD |
2.7 |
erlotinib, cetuximab, bevacizumab |
SD |
4.1 |
5 |
Adenocarcinoma |
PIK3CA: Not done KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
SD |
4.4 |
6 |
Adenocarcinoma |
PIK3CA: No KRAS: No TP53: Not done |
Yes |
SD |
6.2 |
erlotinib, cetuximab, bevacizumab |
SD |
9.2 |
7 |
Adenocarcinoma |
PIK3CA: Not done KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
PD |
2.0 |
8 |
Adenocarcinoma |
PIK3CA: Not done KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
PD** |
3.8 |
9 |
Adenocarcinoma |
PIK3CA: No KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
SD |
6.5 |
10 |
Adenocarcinoma |
PIK3CA: No KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
SD |
11.0 |
11 |
Adenocarcinoma |
PIK3CA: Not done KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
SD |
2.2 |
12 |
Adenocarcinoma |
PIK3CA: Not done* KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
PR |
4.1 |
13 |
Adenocarcinoma |
PIK3CA: No KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
SD |
3.3 |
14 |
Squamous cell carcinoma |
PIK3CA: Not done KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
PD |
2.1 |
15 |
Adenocarcinoma |
PIK3CA: E545K (exon 9) KRAS: No TP53: Not done* |
No |
NA |
NA |
erlotinib, cetuximab, bevacizumab |
SD |
3.1+ |
16 |
Adenocarcinoma |
PIK3CA: Not done KRAS: No TP53: Not done |
No |
NA |
NA |
erlotinib, cetuximab |
SD |
2.0 |
17 |
Squamous cell carcinoma |
PIK3CA: No KRAS: Not done* TP53: Not done |
Yes |
SD |
6.0 |
erlotinib, cetuximab |
too early |
1.5+ |
18 |
Adenocarcinoma |
PIK3CA: No KRAS: No TP53: Not done |
Yes |
PD** |
0.7 |
erlotinib, cetuximab |
too early |
1.6+ |
19 |
Adenocarcinoma |
PIK3CA: No KRAS: No TP53: V157F (exon 5) |
Yes |
PD** |
0.9 |
erlotinib, cetuximab |
PD** |
0.5 |
20 |
Adenocarcinoma |
PIK3CA: No KRAS: G12D TP53: Not done* |
Yes |
PD |
2.3 |
erlotinib, cetuximab |
too early |
0.0+ |
21 |
Adenocarcinoma |
PIK3CA: No KRAS: G12D TP53: Not done* |
No |
NA |
NA |
erlotinib, cetuximab |
too early |
0.1+ |
22 |
Adenocarcinoma |
PIK3CA: No KRAS: Not done TP53: Not done |
Yes |
PD** |
0.8 |
erlotinib, bortezomib |
PD** |
0.3 |
23 |
Neuroendocrine |
PIK3CA: E545K (exon 9) KRAS: Not done TP53: Not done |
No |
NA |
NA |
erlotinib, dasatinib |
SD |
2.8 |
24 |
Adenocarcinoma |
PIK3CA: Not done KRAS: No TP53: Not done |
Yes |
PD |
2.0 |
cetuximab, sirolimus |
too early |
0.9+ |
Abbreviations: EGFR, Epidermal Growth Factor Receptor; KRAS, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; NA, Not applicable; NSCLC, Non-Small Cell Lung Cancer; PR, Partial response; PIK3CA, Phosphatidylinositol 3-kinase, catalytic, alpha polypeptide; SD, Stable disease; PD, Progressive disease; TTF, Time to treatment failure; TP 53, Tumor Protein p53
*not done because tissue was not available for molecular analysis
**clinical progression/new metastasis
+did not progress at the time of analysis
KRAS/TP53:
The two EGFR wild-type patients with a KRAS mutation (cases #20 and 21, Table 3) were not evaluable as they had not reached the post-treatment assessment. None of the 19 evaluable patients with EGFR wild-type had a KRAS mutation. Of the two EGFR wild-type patients with a TP53 mutation, one patient (case #1, Table 3) had prolonged SD for 26.5 months on erlotinib/cetuximab/bevacizumab; the other patient (case #19, Table 3) had PD after 0.5 months on erlotinib/cetuximab.
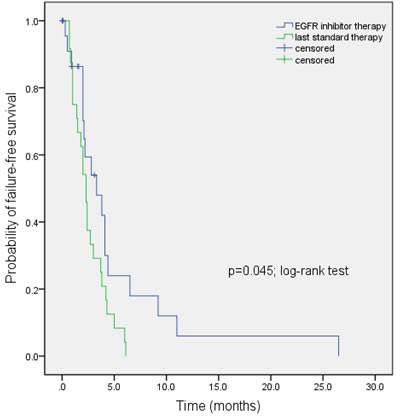
Figure 3: Kaplan-Meier estimates of time to treatment failure in 24 NSCLC patients with EGFR wild-type disease treated with EGFR inhibitor-based combination therapies in phase I clinical trial program (3.3 months) vs. time to treatment failure on their last standard therapy (2.3 months; p=0.045; log-rank test)
Overall Survival
The median OS of 15 EGFR mutation-positive patients treated on EGFR inhibitor-based regimens from the date of start of therapy was 4.7 months (95% CI, 3.5 – 5.9 months).The one year survival rate was 31% (95% CI, 22.3-41.1%). At the time of analysis, 12 of 15 patients were dead. The median OS of 24 EGFR wild-type patients treated on the same regimens was 3.8 months (95% CI, 0.6-7.0 months).
DISCUSSION
The identification of molecular aberrations and selection of therapy to ‘match’ these aberrations is gaining momentum as a preferred treatment approach[14-24]. In the Phase I setting, studies have sought to validate this approach across a range of mutation types, and to support broad genomic testing.
EGFR mutations, frequently observed in patients with NSCLC, activate the kinase activity of EGFR, leading to upregulation of downstream survival pathways[25, 26]. In our study, EGFR mutations in exons 18-21 were present in 21 of 131 patients (16%) with NSCLC. Despite the dramatic initial responses to single-agent EGFR tyrosine kinase inhibitors (TKIs) in EGFR mutation-positive NSCLC patients, 70% of them relapse within one year of initiation of therapy[7, 8, 27]. It has been reported that kinase-independent activity of EGFR prevents autophagic cell death, perhaps accounting for progression of disease despite treatment with tyrosine kinase inhibitors [10]. It is conceivable that combining EGFR kinase inhibitors with EGFR antibody may therefore overcome resistance. Based on preclinical studies, other strategies to augment EGFR inhibition includes use of bortezomib in combination with EGFR kinase inhibitors due to its growth inhibitory and pro-apoptic effects on cancer cell lines[12] and dasatinib in combination with EGFR kinase inhibitor as in vitro data from NSCLC cell lines demonstrates that Src inhibition may enhance the antitumor activity of EGFR inhibition in the presence of EGFR mutations[11].
In our study, eight of 34 evaluable patients (23%) treated with EGFR inhibitor-based combinations achieved SD ≥6 months/PR. Those individuals include EGFR mutation-positive patients with a de novo resistant mutation (n=1); EGFR mutation-positive (sensitive mutation) and prior secondary resistance to EGFR inhibitors (n=2); and, EGFR wild-type disease (n=5; including a patient with squamous cell histology).
Of the 15 EGFR mutation-positive NSCLC patients treated on EGFR inhibitors, three (20%) had SD ≥6 months/PR (cases #2, 5 and 10, Table 2). One patient with a known EGFR-resistant mutation (insertion in exon 20; case #2, Table 2) has an ongoing PR (33% decrease) at 13+ months on therapy with erlotinib and the EGFR antibody cetuximab. Indeed, there is preclinical data demonstrating superiority when EGFR kinase inhibitors are combined with EGFR antibody in regard to antitumor effect[28, 29]
A second patient (case #5, Table 2) with two known EGFR-sensitive mutations (L858R and G873E in exon 21) attained a PR (55% decrease) for 9+ months on erlotinib/cetuximab/bevacizumab. Interestingly, this patient also had a PIK3CA mutation (E542K in exon 9) in the downstream signaling pathway which is a known resistant mechanism to EGFR inhibition[30]. The PIK3CA mutation may explain why the patient had previously developed secondary resistance to single-agent erlotinib. Preclinical studies have demonstrated antitumor activity with PI3K/mTOR inhibitor combinations in gefitinib-resistant PIK3CA-mutant NSCLC cell lines[31]. Clinical trials with dual blockade of PI3K and mTOR are underway[32, 33]. The response seen in this patient on erlotinib/cetuximab/bevacizumab may be due to synergistic effect of simultaneous EGFR and vascular endothelial growth factor (VEGF) inhibition, as demonstrated in preclinical models, including EGFR inhibitor-resistant cell lines[34] and in patients with metastatic colorectal cancer[35], and NSCLC[36, 37]. A third patient (case #10, Table 2) with a known EGFR-sensitive mutation (L858R), with previous response to single agent erlotinib and subsequent resistance, has ongoing SD (26% decrease) for 10+ months on erlotinib and bortezomib. There is preclinical evidence that dual blockade of proteasome activity and EGFR function by combining bortezomib plus cetuximab has synergistic antitumor activity[12]. Patients treated with erlotinib may also benefit from re-treatment[38], so this could also explain the observation of prolonged stable disease, at least in part. However, the TTF on prior erlotinib was 5.5 months, while the current TTF is 10+ months, suggesting that the longer current TTF could not be due to erlotinib alone. These observations indicate that combining treatment with drugs that target different signaling pathways may help patients who had progressed on single-agent targeted therapy after a period of initial response.
Unexpectedly, patients with EGFR wild-type disease were also noted to have salutary effects on EGFR inhibitor-based combination therapies. Five of 19 evaluable NSCLC patients with wild-type EGFR treated on the same EGFR inhibitor-based regimens attained SD ≥6 months/PR. The median TTF on EGFR inhibitor-based regimen (3.3 months) was significantly longer than the median TTF on their last standard therapy (2.3 months; p=0.045; Figure 3). These data suggest that EGFR-based combinations can be active in patients with wild-type disease[39, 40]. All these patients received erlotinib/cetuximab/bevacizumab. Previously, modest antitumor activity has been reported in 4 of 13 NSCLC patients with wild-type EGFR (31%) on gefitinib and cetuximab[41].
We also noted anecdotal activity in patients with squamous cell histology. Of the three evaluable patients with squamous cell histology who were treated with EGFR inhibitor-based regimens, two patients had EGFR wild-type disease (cases #1 and 14, Table 3) and one patient was EGFR-mutant (case #15, Table 2). One patient (TP53-mutation positive; case #1, Table 3) with EGFR wild-type, squamous cell histology achieved SD for 26.5 months on erlotinib/cetuximab/bevacizumab after progression on two prior therapies. Limited data exists regarding patients with squamous cell histology treated with EGFR inhibitors because these patients typically have EGFR wild-type disease[42]. In one study [43], 121 patients with squamous cell carcinoma of the lung were treated with single-agent erlotinib. Thirty seven of 69 evaluable patients achieved PR/SD; however, the duration of response or molecular aberrations was unknown. In another study[44], 1,125 NSCLC patients including 190 patients with squamous cell carcinoma were randomized to either chemotherapy alone or to cetuximab plus chemotherapy. A survival advantage was observed in 557 NSCLC patients treated with cetuximab plus chemotherapy; however, information on which of these responders had squamous cell histology was not reported. Finally, one responsive patient was noted to have a TP53 mutation. Recent retrospective data analysis posits longer progression-free survival for TP53-mutant patients on bevacizumab-based regimens[45].
The activity noted in these subtypes of NSCLC is hypothesis-generating; however, sample size is a significant limitation of this study. The heterogeneity of combination treatments and the retrospective nature of the analysis must also be considered. Therefore, any interpretation of these results needs to be approached with caution. Furthermore, the antitumor activity seen in two patients with EGFR-mutant NSCLC (cases #5 and 10, Table 2), who had progressed on prior treatment with erlotinib after initial response, may be due to the re-treatment effect that occurs in patients with EGFR mutant disease with reintroduction of an EGFR tyrosine kinase inhibitor after a drug holiday[38]. However, in the latter case (case #10, Table 2), the TTF is ongoing and at least double the TTF on prior EGFR inhibitor therapy.
In conclusion, this study demonstrated that treatment with EGFR inhibitor-based combination therapies was associated with SD ≥6 months/PR in subtypes of heavily pretreated advanced NSCLC not traditionally associated with response to EGFR inhibitors, including 1 of 2 patients with a de novo EGFR-resistant mutation; 2 of 12 patients (17%) with an EGFR-sensitive mutation and secondary EGFR resistance after a period of initial response; 5 of 19 evaluable patients (26%) with EGFR wild-type disease; and, 1 of 3 evaluable patients (25%) with squamous cell carcinoma. Further exploration of rational EGFR inhibitor combinations in a broad range of patients with NSCLC may be warranted.
METHODS
Patients
We investigated the EGFR mutation status of 131 consecutive patients with NSCLC referred to the Department of Investigational Cancer Therapeutics (Phase I Clinical Trials Program) at The University of Texas MD Anderson Cancer Center (MDACC) beginning January 1, 2009. The study and all treatments were conducted in accordance with the guidelines of the MD Anderson Institutional Review Board.
Tissue samples and mutation analyses
EGFR mutations were investigated in archival formalin-fixed, paraffin-embedded tissue blocks or material from fine needle aspiration biopsy obtained from diagnostic and/or therapeutic procedures. All histologies were centrally reviewed at MDACC. EGFR mutation testing was done in the Clinical Laboratory Improvement Amendment–certified Molecular Diagnostic Laboratory within the Division of Pathology and Laboratory Medicine at MDACC.
DNA was isolated from formalin-fixed, paraffin-embedded tissue by using a QIAmp DNA Minikit (Qiagen Inc., Valencia, CA) according to the manufacturer’s instructions. EGFR exons 18-21 sequence were analyzed in both sense and antisense directions for the presence of mutations using nested PCR followed by direct sequencing of the nested PCR amplicons. The nested-PCR was done using the primers and annealing conditions as described by Lynch et al[3]. The nested PCR amplicons were purified using the Qiagen QIAquick PCR Purification Kit, followed by cycle-sequencing using BigDye Terminator Kit v1.1 (ABI, Foster City, CA) on ABI Prism 3130 Genetic Analyzer, according to manufacturer’s instructions. Whenever possible, in addition to EGFR, we tested for other mutations such as PIK3CA (codons 532 to 554 in exon 9 and codons 1011 to 1062 in exon 20), KRAS (codons 12, 13, and 61) and TP53 (exons 4 to 9).
Treatment and evaluation
EGFR inhibitors included the small molecule inhibitor erlotinib and the antibody cetuximab. Rationale for the trials was based on preclinical work demonstrating synergistic or additive effects and/or clinical work suggesting complementary pathway inhibition. The trials included erlotinib/cetuximab/bevacizumab (ClinicalTrials.gov identifier: NCT00543504); erlotinib/cetuximab (ClinicalTrials.gov identifier: NCT00895362); erlotinib/bortezomib (ClinicalTrials.gov identifier: NCT00895687); erlotinib/dasatinib (ClinicalTrials.gov identifier: NCT00895128); and cetuximab/sirolimus (ClinicalTrials.gov identifier: NCT00940381). Treatment was chosen based on trial availability and physician and patient preference. Treatment continued until disease progression or unacceptable toxicity occurred. Treatment was carried out according to the specific requirements of the treatment protocols selected. Assessments, including history, physical examination, and laboratory evaluations, were done as specified in each protocol, typically before the initiation of therapy, weekly during the first cycle, and then, at a minimum, at the beginning of each new treatment cycle.
Response assessment
Efficacy was assessed from computed tomography (CT) scans and/or magnetic resonance imaging (MRI) and/or positron emission tomography (PET) scan at baseline before treatment initiation and then every 2-3 cycles (6–12 weeks), depending on the protocol. All radiographs were read in the Department of Radiology at MDACC and reviewed in the Department of Investigational Cancer Therapeutics tumor measurement clinic. Responses were categorized per Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 [46] criteria and were reported as best response. Complete response (CR) was defined as the disappearance of all measurable and non-measurable disease; partial response (PR) was defined as at least a 30% decrease in the sum of the longest diameter of measurable target lesions; progressive disease (PD) was defined as at least a 20% increase in the sum of the longest diameter of measurable target lesions, or unequivocal progression of a non-target lesion, or the appearance of a new lesion; and stable disease (SD) was defined as neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD. A waterfall plot was used to illustrate the anti-tumor activity observed in patients evaluable for response.
Statistical analysis
Patient characteristics, including demographics, EGFR mutation status and prior treatment with an EGFR inhibitor were summarized using frequencies and percentages. Time to treatment failure (TTF) was defined as the time interval between the start of therapy and the date of disease progression or death, whichever occurred first. Patients who were alive and had not failed treatment were censored at the time of their last follow-up. The Kaplan-Meier method [47] was used to estimate TTF and log-rank tests [48] were performed to compare subgroups of patients. All tests were two-sided, and P <0.05 was considered statistically significant. All statistical analyses were carried out using SPSS (version 19.0; SPSS, Chicago, IL, USA).
Disclosures
Dr. Razelle Kurzrock received honoraria and research funding from Genetech. The remaining authors declare no conflicts of interest.
References
1. Sharma SV, Bell DW, Settleman J and Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007; 7(3):169-181.
2. Chintala L and Kurzrock R. Epidermal growth factor receptor mutation and diverse tumors: case report and concise literature review. Mol Oncol. 2010; 4(4):306-308.
3. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J and Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350(21):2129-2139.
4. Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M and Miller VA. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006; 12(3 Pt 1):839-844.
5. Janku F, Garrido-Laguna I, Petruzelka LB, Stewart DJ and Kurzrock R. Novel therapeutic targets in non-small cell lung cancer. J Thorac Oncol. 2011; 6(9):1601-1612.
6. Okamoto I, Araki J, Suto R, Shimada M, Nakagawa K and Fukuoka M. EGFR mutation in gefitinib-responsive small-cell lung cancer. Ann Oncol. 2006; 17(6):1028-1029.
7. Hammerman PS, Janne PA and Johnson BE. Resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2009; 15(24):7502-7509.
8. Mumenthaler SM, Foo J, Leder K, Choi NC, Agus DB, Pao W, Mallick P and Michor F. Evolutionary modeling of combination treatment strategies to overcome resistance to tyrosine kinase inhibitors in non-small cell lung cancer. Mol Pharm. 2011; 8(6):2069-2079.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005; 353(2):123-132.
10. Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ and Hung MC. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008; 13(5):385-393.
11. Song L, Morris M, Bagui T, Lee FY, Jove R and Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006; 66(11):5542-5548.
12. Cascone T, Morelli MP, Morgillo F, Kim WY, Rodolico G, Pepe S, Tortora G, Berrino L, Lee HY, Heymach JV and Ciardiello F. Synergistic anti-proliferative and pro-apoptotic activity of combined therapy with bortezomib, a proteasome inhibitor, with anti-epidermal growth factor receptor (EGFR) drugs in human cancer cells. J Cell Physiol. 2008; 216(3):698-707.
13. Yasuda H, Kobayashi S and Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012; 13(1):e23-31.
14. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S and Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001; 344(14):1031-1037.
15. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347(7):472-480.
16. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K and Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363(9):809-819.
17. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O’Day SJ, Blackman SC, Curtis CM, Lebowitz P, Ma B, Ouellet D, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012; 379(9829):1893-1901.
18. Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, Shapiro GI, Costa DB, Ou SH, Butaney M, Salgia R, Maki RG, Varella-Garcia M, Doebele RC, Bang YJ, Kulig K, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011; 12(11):1004-1012.
19. Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, Wheler JJ, Moulder SL, Fu S and Kurzrock R. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011; 10(3):558-565.
20. Braiteh F and Kurzrock R. Uncommon tumors and exceptional therapies: paradox or paradigm? Mol Cancer Ther. 2007; 6(4):1175-1179.
21. Garrido-Laguna I, Hidalgo M and Kurzrock R. The inverted pyramid of biomarker-driven trials. Nat Rev Clin Oncol. 2011; 8(9):562-566.
22. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH and Kurzrock R. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30(8):777-782.
23. Moroney J, Wheler J, Hong D, Naing A, Falchook G, Bodurka D, Coleman R, Lu KR, Xiao LC and Kurzrock R. Phase I clinical trials in 85 patients with gynecologic cancer: The M. D. Anderson Cancer Center experience. Gynecologic Oncology. 2010; 117(3):467-472.
24. Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Luthra R, Ye Y, Wen S, Berry D and Kurzrock R. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012; 18(22):6373-6383.
25. Sordella R, Bell DW, Haber DA and Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004; 305(5687):1163-1167.
26. Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M and Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005; 65(19):8968-8974.
27. Sequist LV, Bell DW, Lynch TJ and Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007; 25(5):587-595.
28. Huang S, Armstrong EA, Benavente S, Chinnaiyan P and Harari PM. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 2004; 64(15):5355-5362.
29. Matar P, Rojo F, Cassia R, Moreno-Bueno G, Di Cosimo S, Tabernero J, Guzman M, Rodriguez S, Arribas J, Palacios J and Baselga J. Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC-C225): superiority over single-agent receptor targeting. Clin Cancer Res. 2004; 10(19):6487-6501.
30. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011; 3(75):75ra26.
31. Zou ZQ, Zhang XH, Wang F, Shen QJ, Xu J, Zhang LN, Xing WH, Zhuo RJ and Li D. A novel dual PI3Kalpha/mTOR inhibitor PI-103 with high antitumor activity in non-small cell lung cancer cells. Int J Mol Med. 2009; 24(1):97-101.
32. Pao W and Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011; 12(2):175-180.
33. Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, Zakowski MF, Rusch V, Sima CS, Ladanyi M and Kris MG. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther. 2012; 11(2):485-491.
34. Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol Cancer Res. 2007; 5(3):203-220.
35. Saltz LB, Lenz HJ, Hochster H, Wadler S, Hoff P, Kemeny N, Hollywood E, Gonen M, Wetherbee S and Chen H. Randomized phase II trial of cetuximab/bevacizumab/irinotecan (CBI) versus cetuximab/bevacizumab (CB) in irinotecan-refractory colorectal cancer. Journal of Clinical Oncology. 2005; 23(16):248s-248s.
36. Herbst RS, Johnson DH, Mininberg E, Carbone DP, Henderson T, Kim ES, Blumenschein G, Jr., Lee JJ, Liu DD, Truong MT, Hong WK, Tran H, Tsao A, Xie D, Ramies DA, Mass R, et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005; 23(11):2544-2555.
37. Janku F, Stewart DJ and Kurzrock R. Targeted therapy in non-small-cell lung cancer--is it becoming a reality? Nat Rev Clin Oncol. 2010; 7(7):401-414.
38. Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, Lynch T, Johnson BE and Miller VA. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010; 28(2):357-360.
39. Wang F, Wang J, Bai H, Zhao J, Wang Z, Zhuo M, An T, Duan J, Yang L, Wu M, Wang Y, Wang S and Liu X. An evaluation of phosphorylated EGFR expression in predicting outcome of EGFR-TKI therapy for the advanced NSCLC patients with EGFR wild type [abstract 7532]. J Clin Oncol 2011; 29:s 2011.
40. Falchook GS, Naing A, Hong DS, Zinner R, Fu S, Piha-Paul SA, Tsimberidou AM, Morgan-Linnell SK, Jiang Y, Bastida C, Wheler JJ and Kurzrock R. Dual EGFR inhibition in combination with anti-VEGF treatment: A phase I clinical trial in non-small cell lung cancer. Oncotarget. 2013; 4(1):118-127.
41. Ramalingam S, Forster J, Naret C, Evans T, Sulecki M, Lu H, Teegarden P, Weber MR and Belani CP. Dual inhibition of the epidermal growth factor receptor with cetuximab, an IgG1 monoclonal antibody, and gefitinib, a tyrosine kinase inhibitor, in patients with refractory non-small cell lung cancer (NSCLC): a phase I study. J Thorac Oncol. 2008; 3(3):258-264.
42. Rekhtman N, Paik PK, Arcila ME, Tafe LJ, Oxnard GR, Moreira AL, Travis WD, Zakowski MF, Kris MG and Ladanyi M. Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clin Cancer Res. 2012; 18(4):1167-1176.
43. Gurpide A, Massuti B, Pallares C, Salinas P, Montes A, Lopez-Vivanco G, Baron MG, Cobo M, Amador ML and Paz-Ares L. Erlotinib in patients with advanced squamous cell carcinoma of the lung. J Clin Oncol. 2006; 24(18):407s-407s.
44. Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, Vynnychenko I, Park K, Yu CT, Ganul V, Roh JK, Bajetta E, O’Byrne K, de Marinis F, Eberhardt W, Goddemeier T, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009; 373(9674):1525-1531.
45. Said R, Hong D, Wheler J, Naing A, Falchook G, Fu S, Janku F, Zinner R, Piha-Paul S, Warneke CL, Tsimberidou A and Kurzrock R. (2012). p53 mutations in advanced cancers: clinical characteristics and outcomes in a phase I setting. J Clin Oncol 30, 2012 (suppl; abstr 10607)
46. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC and Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000; 92(3):205-216.
47. Kaplan EL and Meier P. Nonparametric estimation from incomplete observations Journal of the American Statistical Association 1958; 53(282):457-481.
48. Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966; 50(3):163-170.