
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is the most common pancreatic tumor (accounting for more than 90% of cases). PDA is aggressive and difficult to detect at an early stage, with few effective treatments and low survival rates, making it a major challenge for biomedical research. Indeed, this adenocarcinoma is predicted to become the second leading cause of cancer-related deaths in the United States by 2020 [1, 2]. Current therapies for unresectable tumors, which are the most common ones, include chemotherapy administration based on gemcitabine, folifirinox or nab-paclitaxel, but these are very inefficient and only minimally improve patient survival, such that new therapeutic strategies with improved efficiency are urgently needed. In contrast, other types of less-frequent pancreatic tumors, such as acinar cell carcinomas [3] and pancreatic neuroendocrine tumors [4], are associated with better prognosis and longer survival rates.
Sunitinib is a broad-spectrum receptor tyrosine kinase (RTK) inhibitor whose targets include VEGFR-1,-2,-3, PDGFR-α,-β [5], stem cell factor receptor (c-KIT), Fms-like tyrosine kinase-3 receptor, the glial cell line derived neurotrophic factor receptor (RET) and colony-stimulating factor type 1 receptor (CSF-1R) [5–7]. Sunitinib inhibits endothelial cell proliferation and therefore is considered as an anti-angiogenic drug [8]. However, the expression of sunitinib-targeted RTKs in other stromal cells, as well as in the tumor epithelium, suggests that it could have a more extended mechanism of action with potential multiple effect in these different cells [5, 9, 10]. Sunitinib treatment has given impressive results in neuroendocrine pancreatic tumors, and its use for this pathology was recently approved by the FDA [11, 12]. In contrast, phase II clinical trials to test sunitinib for treating PDA have consistently shown its failure, either when sunitinib was combined with gemcitabine for patients with advanced or metastatic PDA [13] or when it was given as a second-line therapy after gemcitabine failure [14].
The reasons behind this inefficacy of sunitinib treatment in PDA patients are not well understood. Several in vitro and in vivo reports on pancreatic ductal tumor cell lines and xenografts showed encouraging results, suggesting that sunitinib could potentially be used to improve standard chemotherapeutical treatments [10, 15–20]. In vivo sunitinib treatment in transgenic mouse models of pancreatic cancer was ineffective for k-ras–driven PDA yet successful for a neuroendocrine preclinical model (RIP-Tag) [21]. Understanding these apparently contradictory results thus requires further studies to elucidate the precise mechanistic roles and potential uses of sunitinib in PDA therapy.
Here, we have tested sunitinib effects in the Ela-myc transgenic mouse model of pancreatic cancer, both in vitro and in vivo. Ela-myc mice develop acinar tumors that can undergo acinar-to-ductal metaplasia and progress to ductal adenocarcinomas, mimicking human PDA progression [22–24]. In contrast to xenograft models, transgenic mice can better recapitulate tumor onset, tumor progression and tumor-stroma crosstalk, the latter of which is particularly important in pancreatic cancer. This study using the Ela-myc model complements the only one previously published preclinical report of sunitinib in pancreatic cancer that used transgenic mice (with a k-ras–based model) [21]. Using the Ela-myc model, we were able to analyze sunitinib effects in ductal as well as, for the first time, in acinar tumors. Our in vivo and in vitro data provide new insights into the therapeutic use of sunitinib for exocrine pancreatic cancer.
RESULTS
Ela-myc tumors express sunitinib-targeted tyrosine kinase receptors in both stromal and cancer cells
The Ela-myc transgenic mouse model overexpresses the oncogene c-myc under the control of the elastase promoter, which leads to acinar pancreatic tumors. Importantly, approximately 50% of these tumors progress to ductal tumors, which are associated with abundant stroma—one of the principle hallmarks of human PDA [22, 23]. Although mutation of K-Ras is the most frequent alteration driving pancreatic cancer, c-Myc appears to have a key role in PDA development and progression [25–27] and has also been linked with K-Ras in this pathology [28, 29]. To test the effects of sunitinib in this model of pancreatic cancer, we first analyzed the expression of sunitinib-targeted RTK in acinar and ductal tumor areas from Ela-myc mice (Figure 1). Immunohistochemical analyses showed that VEGFR2, PDGFR-α and PDGFR-β were expressed in pancreatic tumors, both acinar (Figure 1a–1f) and ductal areas (Figure 1g–1l). Importantly, these receptors were expressed not only in blood vessels and tumor stroma (Figure 1, arrows) but also in epithelial cancer cells (Figure 1, arrowheads). These data suggested a potential sunitinib sensitivity of Ela-myc tumors, which could be due not only to its well-known anti-angiogenic effect but also to its effects on the RTK that are expressed in pancreatic tumor cells.
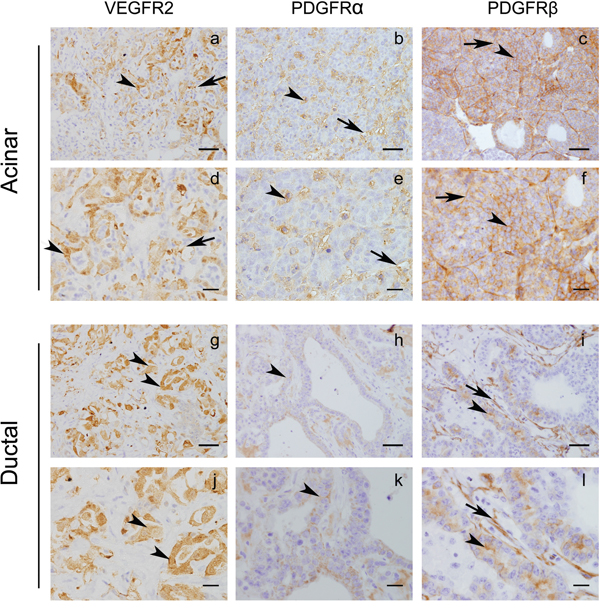
Figure 1: Ela-myc tumors express tyrosine kinase receptors that can be targeted by sunitinib. Immunohistochemical analyses of Ela-myc mice pancreatic tumors were performed to detect tyrosine kinase receptors known to be sunitinib targets, such as VEGFR2, PDGFR-α and PDGFR-β. Scale bars for a–c. and g–i., 50 μm; for d–f. and j–l., 20 μm.
Sunitinib treatment effects on survival, tumor burden, differentiation and necrosis in Ela-myc mice
Next, we analyzed the effects of sunitinib treatment in tumor progression and survival of Ela-myc mice. These mice develop pancreatic cancer with 100% penetrance between 2 and 7 months of age. Ela-myc mice with advanced tumors (4.5 months old) were treated by oral administration of sunitinib or control vehicle for 15 days. After this treatment, animals were followed until endpoint disease as determined by strict ethical guidelines (Figure 2A). Sunitinib treatment had no significant effect on Ela-myc mice survival, as determined by Kaplan-Meier curve analyses (Figure 2B). Similarly, Ela-myc tumors from sunitinib-treated mice did not display differences in tumor mass (Figure 2C). Finally, further histopathologic analyses of tumor samples to evaluate tumor differentiation, as determined by the percentage of acinar versus ductal component in each tumor (Figure 2D) and by necrosis (Figure 2E), failed to detect significant differences between sunitinib-treated and control mice. These data indicate that sunitinib therapy is inefficient in well-established pancreatic tumors in the Ela-myc cancer model.
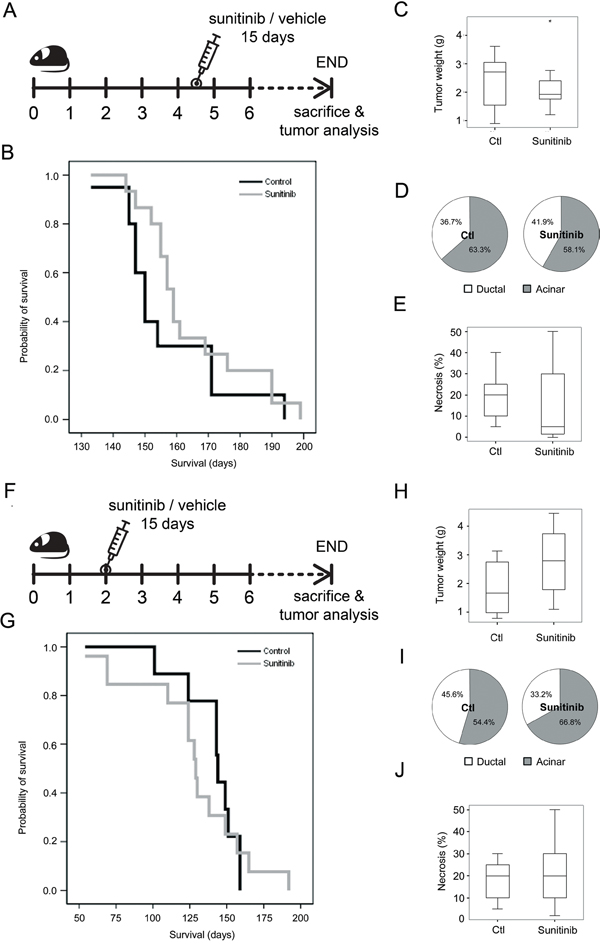
Figure 2: The effects of sunitinib administration on tumor progression and survival in the Ela-myc mice model. A. Ela-myc mice with well-established pancreatic tumors (4.5-months old) were treated daily with sunitinib (80 mg/kg; n = 20) or control vehicle (n = 20) for 15 days. Mice were sacrificed at endpoint based on ethical guidelines to study animal survival. B. A Kaplan-Meier survival plot showing no significant differences between control or sunitinib-treated animals. C. Box-and-whisker plot showing tumor weight at the moment of sacrifice. The asterisk present in the sunitinib treated group is not referring to significance but indicating an extreme outlier. D. Pie charts showing the percentage of ductal (white) and acinar (grey) areas in control or sunitinib-treated animal. E. Box-and-whisker plot showing tumor necrosis in acinar areas, as determined histopathologically. F. Sunitinib administration scheme in 2-month-old Ela-myc mice harboring incipient tumors. Similar to treatment of 4.5-month-old mice, no statistically significant differences were observed between treated and control mice regarding survival G., tumor weight H., tumor differentiation I. or necrosis J.
To determine if mice at earlier stages of pancreatic tumorigenesis were sensitive to sunitinib therapy, 2-month-old Ela-myc mice (which usually have incipient tumors) were treated with sunitinib in a similar manner as the older mice (Figure 2F). However, sunitinib treatment did not significantly affect animal survival (Figure 2G) tumor weight (Figure 2H), differentiation (Figure 2I) or necrosis (Figure 2J).
Sunitinib treatment effects on tumor angiogenesis in Ela-myc mice
As sunitinib treatment inhibits blood vessel formation in cancer, we tested for an anti-angiogenic effect on Ela-myc mice by analyzing the number of blood vessels in pancreatic tumors using von Willebrand factor immunohistochemical staining (Figure 3). Ela-myc pancreatic ductal tumor areas displayed higher vascularization as compared to acinar ones (Figure 3; compare a with c), suggesting increased susceptibility to sunitinib anti-angiogenic effects. However, quantification of the number of blood vessels did not show any differences between vehicle-treated (control) and sunitinib-treated mice, either in acinar or ductal tumor areas (Figure 3, right panel). These data indicate that Ela-myc pancreatic tumors are not responsive to the anti-angiogenic effects of sunitinib.
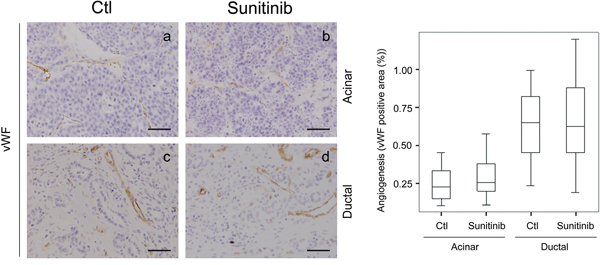
Figure 3: Sunitinib effects on Ela-myc pancreatic tumor angiogenesis. Tumor angiogenesis was evaluated for pancreatic tumors in control (Ctl) and sunitinib-treated Ela-myc mice by von Willebrand Factor (vWF) staining, in both acinar and ductal regions. Quantification of positive areas is shown on the right. Scale bars, 50 μm.
Sunitinib treatment effects on cancer cells in Ela-myc mice
Considering the high expression of sunitinib-targeted RTKs found in Ela-myc pancreatic cancer cells (Figure 1), we next aimed to determine whether sunitinib treatment may directly impact cancer cells, by either inducing apoptosis or blocking tumor cell proliferation. To evaluate sunitinib effects on cancer cell apoptosis, we analyzed cleaved caspase 3 staining of both acinar and ductal tumor areas (Figure 4A). As previously reported [30], acinar tumors showed increased cell apoptosis compared to ductal ones (Figure 4A, compare a with c), although quantification of cleaved caspase 3 positive areas showed no significant differences between control and sunitinib-treated animals (Figure 4A, right panel). Subsequently, we tested whether sunitinib treatment could inhibit tumor cell growth by measuring cell proliferation using P-Histone H3 for acinar tumors (Figure 4B, a and b) and Ki67 immunostaining for ductal tumors (Figure 4B, c and d) (see Material and Methods). No significant differences in the proliferative rate of acinar or ductal pancreatic tumor lesions were detected between vehicle-treated and sunitinib-treated animals (Figure 4B, right panels).
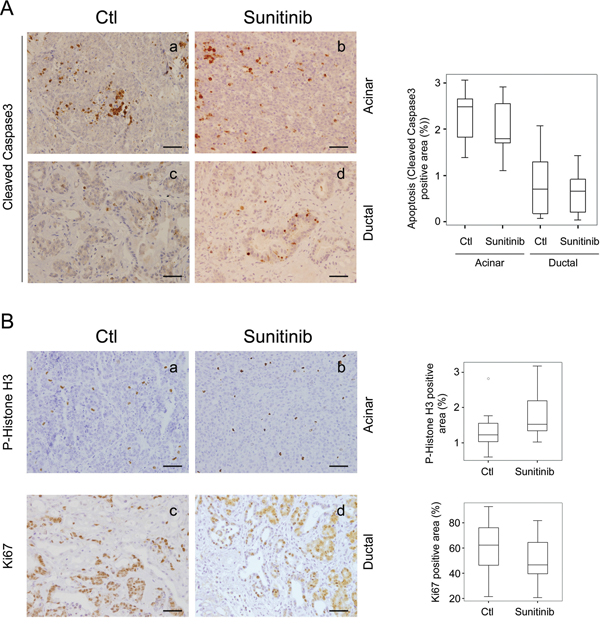
Figure 4: Sunitinib effects in Ela-myc pancreatic tumor cell apoptosis and proliferation. Tumor cell apoptosis and proliferation was analyzed for pancreatic tumors in control (Ctl) and sunitinib-treated Ela-myc mice. A. Tumor cell apoptosis was evaluated through cleaved caspase 3 (active form) staining. B. Tumor cell proliferation was evaluated through P-Histone H3 staining in acinar lesions, or Ki67 in ductal ones. Box-and-whisker plot quantifications are shown on the right (A, B). Scale bars, 50 μm.
Altogether, these data indicate that in vivo sunitinib treatment in the Ela-myc model has no effect on tumor angiogenesis, cell proliferation or apoptosis, with no impact on tumor progression or animal survival.
Sunitinib increases cell death and inhibits cell proliferation in Ela-myc tumor-derived cell lines
Fibrotic stroma is one of the major hallmarks of pancreatic cancer, and its role in hindering efficient drug delivery and promoting therapy resistance is well known [31]. To determine if the lack of effects in vivo of sunitinib treatment in Ela-myc mice could be due to impaired drug delivery because of this pancreatic fibrotic barrier, we tested sunitinib in vitro on two distinct Ela-myc pancreatic tumor–derived cell lines (Emyc-1 and Emyc-10). Similar to Ela-myc pancreatic tumors in vivo, these cells expressed the sunitinib-targeted RTKs VEGFR2, PDGFR-α and PDGFR-β (Figure 5A). Next, we tested the susceptibility of these cells in vitro to different doses of sunitinib. Emyc-1 cells treated with sunitinib (1 to 4 μM) showed impaired viability as compared to vehicle-treated cells, as determined by bright field microscopy (Figure 5B). Kinetic quantification of cell viability by MTT detection clearly showed that in vitro sunitinib treatment of Emyc-1 cells was strongly cytotoxic, in a specific and dose-dependent manner, as compared to vehicle treatment (Figure 5C). We then analyzed whether the sunitinib-induced cytotoxicity was a consequence of increased apoptosis, decreased proliferation, or both, by detecting the active form of caspase 3 and P-Histone H3, respectively. In vitro treatment of Emyc-1 cells with sunitinib resulted in increased apoptosis, shown by immunofluorescence and Western blot detection of cleaved caspase 3 (Figure 5D), and in a dose-dependent reduction of cell proliferation, shown by immunofluorescence staining and quantification of P-Histone H3 (Figure 5E). Similar dose-dependent susceptibility to sunitinib treatment was observed in Emyc-10 cells (data not shown).
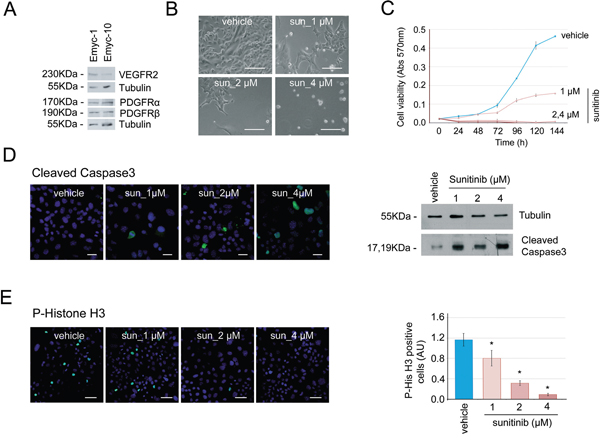
Figure 5: Cells derived from Ela-myc pancreatic tumors are sensitive to sunitinib in vitro. A. Analysis by Western blot of the expresssion of the sunitinib-targeted receptors VEGFR2, PDGFR-α and PDGFR–β in two different cell lines derived from Ela-myc pancreatic tumors. B. Bright field images showing Emyc-1 cell sensitivity to different doses of sunitinib (1, 2 or 4 μM) in vitro, as compared to cells treated with vehicle (DMSO). Scale bars, 100 μm. C. MTT experiments were performed to quantify cell viability upon sunitinib treatment (1, 2 or 4 μM) in the Emyc-1 cell line, observing a dose-dependent effect. D. Left, immunofluorescence of cleaved caspase 3, to detect cell apoptosis upon sunitinib treatment (1, 2 or 4 μM). Scale bars, 20 μm. Right, Western blot analysis of the levels of cleaved caspase 3 in Emyc-1 cell line treated with sunitinib. Tubulin levels are shown as the loading control. E. Left, Immunofluorescence of P-Histone H3, to show cell growth arrest upon sunitinib treatment (1, 2 or 4 μM) in Emyc-1 cells. Scale bars, 50 μm. Right, Bar plots showing quantification of P-Histone H3 immunofluorescence experiments on the right. *p < 0.05 (Student’s t-test).
Thus, in vitro, sunitinib increases cell death and reduces cell proliferation of Ela-myc–derived pancreatic cancer cell lines. This suggests that the lack of effects observed in vivo may be due to impaired drug delivery to the pancreatic tumor, rather than cell insensitivity to the drug.
Sunitinib treatment affects subcutaneous tumors established from Ela-myc tumor-derived cell lines
We next tested the efficiency of sunitinib in vivo on tumors from Emyc-1 cells injected subcutaneously in the dorsal flanks of immunosuppressed mice. Once tumors reached 0.5 cm2, animals were orally treated with sunitinib or control vehicle, and evaluated for drug effects. After a daily treatment with sunitinib or vehicle for 15 days mice were sacrificed and tumors analyzed (Figure 6A). Interestingly, sunitinib administration caused complete tumor regression (Figure 6B, 6C). H&E and histopathological analysis revealed subcutaneous tumors only in mice treated with control vehicle, whereas in mice treated with sunitinib we only observed an inflammatory abscess with no presence of tumor cells (Figure 6D).
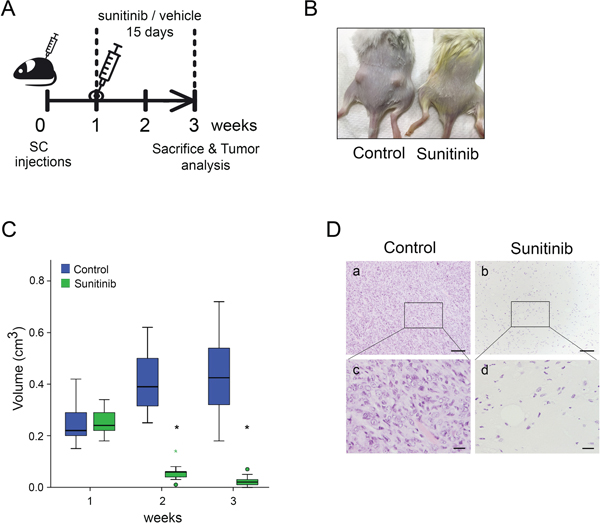
Figure 6: Ela-myc–derived subcutaneous tumors are sensitive to sunitinib. A. Schematic representation of the design of the in vivo subcutaneous experiment. SCID Beige animals were injected with 3 x 106 Emyc-1 cells per flank and, when tumors reached 0.5 cm2 (at 1 week), animals were orally administered with sunitinib or control vehicle for 15 days (10 mice, thus 20 tumors per condition). After treatment, tumors were collected and analysed. B. Representative images of mice with Ela-myc–derived subcutaneous tumors after 15 days of treatment with vehicle or sunitinib. C. Box plot showing tumor size distribution of sunitinib- (green) or vehicle-treated (blue) animals before treatment (week 1), during (week 2) and after (week 3) drug delivery. Green circles label outliers and green asterisks extreme outliers. *p < 0.05 (Mann Whitney test). D. H&E staining of Ela-myc allografts after treatment with control vehicle (panels a, c) or sunitinib (panels b, d). Scale bars, 100 μm (a, b), 20 μm (c, d).
These data suggest that the ineffectiveness of sunitinib treatment in Ela-myc transgenic mice was due to the pancreatic tumor localization. This most likely due to its abundant desmoplastic stroma, as tumors derived from this model in a non-pancreatic niche were highly responsive to the drug in vivo.
DISCUSSION
Gemcitabine was already in use as a first-line PDA therapy by 1997, but without high success rates [32]. Patient survival has slightly improved since then through the use of novel chemotherapy combinations [33, 34], but these new treatments are still unable to reach one year as patient median overall survival, leaving PDA as one of the tumors with the worst prognosis. Thus, new effective therapies are urgently needed to treat this pathology. The multi-target inhibitor of RTKs, sunitinib, recently emerged as a possible hope for PDA therapy. However, although sunitinib therapy has improved treatment of tumors like renal cell cancer and gastrointestinal stromal tumors, it has not led to improvements for PDA patients [13, 14]. Using a preclinical mouse model of pancreatic cancer, we now provide insights to the failure of sunitinib treatment in this pathology as well as potential ways in which this can be overcome. Specifically, we have reported a preclinical study that addresses the impact of sunitinib in pancreatic ductal and acinar tumors using a transgenic mouse model of pancreatic cancer. We found no significant differences on tumor progression or hallmarks or on survival between sunitinib-treated animals and control ones (Figure 7). However, tumor derived cell lines did respond to sunitinib in vitro in a dose-dependent manner. Further, allografts from subcutaneous injections of Ela-myc tumor–derived cells regressed after sunitinib treatment, suggesting that the complex tumor microenvironment present in the pancreatic niche impairs sunitinib effects in the Ela-myc model (Figure 7).
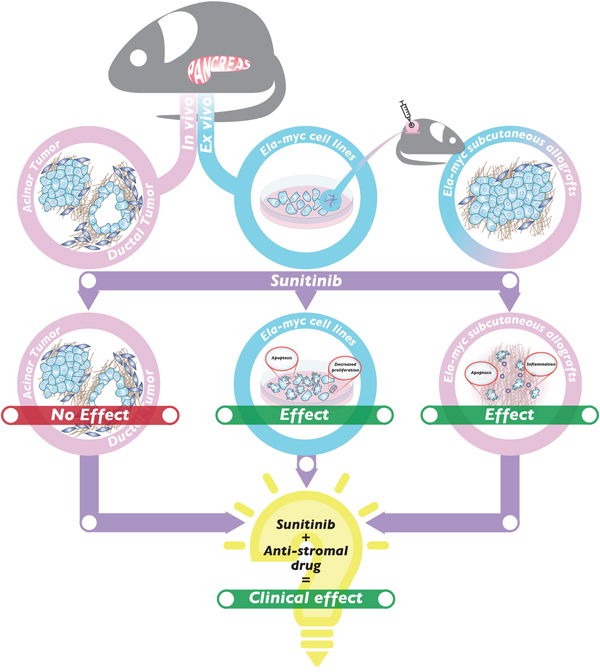
Figure 7: Working model of the in vivo and in vitro effects of sunitinib in the Ela-myc mouse model. Sunitinib treatment of Ela-myc pancreatic acinar and ductal tumors does not show any significant effect in vivo (left). In contrast, Ela-myc–derived tumor cell lines are highly sensitive to sunitinib in vitro, with increased apoptosis and decreased proliferation (center). Importantly, subcutaneous injection of these cells in SCID Beige mice forms tumors that are highly sensitive to sunitinib treatment (right). These data may help understand sunitinib failure for PDA patients and suggest that a combination of sunitinib and an anti-stromal drug may be a promising strategy for successfully treating this pathology in the clinics.
One of the best characterized effects of sunitinib as an anti-cancer therapy is its ability to inhibit angiogenesis by targeting VEGFR on tumor endothelial cells. As angiogenesis is a key contributor to tumor progression and metastasis in most solid tumors, a large number of preclinical and clinical studies have focused on targeting tumor vasculature as an anti-cancer therapy [35]. However, tumor vessels are sparse and poorly functional in PDA, and whether angiogenesis is important for its progression is controversial. On the one hand, hypovascularization of PDA should render these tumors very dependent on this limited vascular network and therefore highly sensitive to anti-angiogenic therapies. Accordingly, several preclinical studies have gathered data showing the benefits of anti-angiogenic treatments in PDA [15, 36, 37]. On the other hand, recent data have shown that increasing blood perfusion and normalizing the vasculature development favor drug delivery and survival in PDA animal models [38]. Moreover, in PDA patients, clinical trials using anti-angiogenic drugs have repeatedly failed [39–41].
An important advantage of sunitinib as compared to standard anti-angiogenic treatments is that it exerts additional anti-tumor effects [42, 43]. Thus, sunitinib can impair tumor desmoplasia by targeting PDGF-dependent fibroblasts proliferation [44] and can block tumor growth by inhibiting several RTKs in cancer cells [5, 6, 42]. Indeed, sunitinib has shown excellent results both in preclinical and clinical settings for different tumors [5, 8, 42, 45, 46] and has gained FDA approval for several neoplasms [11, 47].
However, despite its multi-bullet targeting, sunitinib has not proven effective for PDA. In preclinical mouse models using pancreatic xenografts, the overall survival rate was not improved by treatment of sunitinib alone [10] but was improved when sunitinib was combined with chemotherapy [10, 15–20], radiotherapy [19, 48] or miRNA strategies [49]. In contrast, sunitinib failed to increase survival rates in either the k-ras or the c-myc genetically engineered mouse models ([21] and this manuscript), even in combination with gemcitabine [21]. Here, it is important to note that k-ras– and c-myc–driven pancreatic cancer mouse models, in contrast to xenograft models, recapitulate histopathology and progression of human pancreatic cancer, including acinar-to-ductal metaplasia and a strong desmoplastic reaction [23, 38, 50]. In fact, fibrotic stroma has been reported to act as a mechanical barrier hampering chemotherapy delivery in PDA [31], which could explain the lack of response after sunitinib treatment in these transgenic models. In support of this hypothesis, we found that even though sunitinib treatment had no effects on tumor volume or histopathological hallmarks in Ela-myc mice in vivo, cancer cells derived from these tumors were highly sensitive to the drug in vitro. Further, after injecting cells derived from these tumors into a non-pancreatic niche in the mouse, sunitinib was highly effective in vivo, suggesting that sunitinib exerts its anti-cancer effects if it can efficiently reach the tumor (Figure 7). Importantly, therapeutic targeting of the tumor stroma has given encouraging results in preclinical studies and is now being tested in clinical trials [38, 51, 52]. Moreover, one of the few improvements in managing human PDA in the last 20 years has been by treating with a combination of gemcitabine and nab-paclitaxel, which compromises the stromal architecture to increase perfusion [34]. Preliminary data from mice xenograft PDA models combining nab-paclitaxel with sunitinib are positive [17], although further future preclinical experiments using sunitinib in combination with stromal disrupting drugs are required to better understand the anti-tumor effects of sunitinib in PDA therapy.
In agreement with a lack of sunitinib effectiveness in the k-ras and c-myc genetically engineered mouse models ([21] and this manuscript), sunitinib failed in human PDA treatments when administered as a second-line therapy [14] or in combination with gemcitabine [13]. These data reinforce the idea that xenograft animal models have a low predictive value as compared to genetically engineered models in preclinical studies [53] and highlight the necessity of obtaining extensive preclinical data before moving to patients. In fact, an important meta-analysis of preclinical studies with sunitinib showed that only 2% of these had used genetically engineered mouse models [54]. These data encouraged us to replenish the lack of preclinical information with transgenic models for PDA pathology, using Ela-myc mice as a different transgenic pancreatic tumor model from the previously published k-ras model [21].
The molecular complexity of PDA tumors, which are genetically heterogenous, can explain why sunitinib gave positive results in maintenance therapy in metastatic PDA that had achieved disease control with first-line therapy (a very rare subset of PDA patients) [55]. Likewise, a case report has documented an impressive extended survival after treating with gemcitabine and sunitinib [56]. Thus, the lack of patient selection in published clinical trials may have underestimated the sunitinib potential for PDA treatment, highlighting the importance of personalized medicine in identifying patients who can benefit from sunitinib.
Finally, even though PDA is the most common type of pancreatic cancer, there are patients with other less frequent pancreatic neoplasms who respond poorly to conventional treatments and who might benefit from sunitinib therapy. For instance, patients with pancreatic neuroendocrine tumors, which represent around 1.3% of pancreatic cancers, have shown a very good response to sunitinib in preclinical models [21, 43, 57] and clinical trials [11, 58, 59]. Indeed, sunitinib has been recently approved by the FDA for the treatment of these tumors [11]. These differences in sunitinib response between neuroendocrine and PDA tumors might be explained by the high vascularization of the former and/or by the dense desmoplasic reaction that hampers drug delivery in PDA. Another uncommon pancreatic cancer is acinar cell carcinoma, which accounts for 1% to 2% of all pancreatic tumors in adults and up to 15% in infancy. Surgical resection is the only chance of cure for acinar cancer, and there is a lack of systemic successful therapies for advanced non-resectable tumors [60]. Research on acinar cancer treatment has been handicapped by the low number of acinar cell lines available, which has limited the in vitro laboratory tools, compromised the use of xenografts in vivo and resulted in a lack of preclinical studies addressing acinar tumor therapy. Moreover, as this pancreatic cancer type is very rare [61], clinical trials are unfeasible, limiting studies to small sample sizes and case reports. The transgenic Ela-myc mouse model, which develops mixed acinar and ductal tumors, now provides the first tool for studying sunitinib treatment of pancreatic acinar cell carcinomas. Our data show that, similar to PDA tumors, acinar tumors do not respond in vivo to sunitinib in this preclinical model. Unfortunately, we cannot test in vitro sunitinib effects on acinar cells derived from Ela-myc tumors, as these cells undergo acinar-to-ductal metaplasia in culture. Therefore, whether this lack of sunitinib efficacy in Ela-myc acinar tumors in vivo is due to inefficient drug delivery into the tumor, or to insensitivity of acinar cells to the drug, requires further investigation.
The data reported here fit with previous preclinical results with sunitinib in PDA transgenic models and, more importantly, with unselected clinical trials for this pathology. Overall, these results stress the importance of using genetically engineered mouse models rather than xenograft mouse models in preclinical studies. We hypothesize that combining sunitinib with agents targeting the stroma (rather than with the conventional gemcitabine) and segregating patient cohorts using personalized medicine could result in the successful use of this drug for PDA patients.
MATERIALS AND METHODS
Mice and sunitinib treatments
Animal procedures were approved by the PRBB Ethical Committee for Animal Experimentation. Founder Ela-myc mice (C57Bl/6 genetic background) were kindly provided by E. Sandgren (University of Wisconsin-Madison, WI). Mice were housed and fed ad libitum as previously described [22, 62]. Genotyping primers: c-myc (5′-CAC CGC CTA CAT CCT GTC CAT TCA AGC-3′ and 5′-TTA GGA CAA GGC TGG TGG GCA CTG-3′), resulting in a 200 bp band. SCID Beige mice were obtained from the PRBB Animal Facility. Emyc-1 cells (3 × 106) were injected subcutaneously in the two dorsal lateral flanks of 20 SCID Beige mice. Sunitinib (kindly provided by Pfizer) was prepared in a vehicle solution (0.5% carboxymethylcellulose, 1.8% NaCl, 0.4% Tween-80, 0.9% benzyl alcohol) and kept in the dark. Mice (n = 10 for each condition) were treated for 15 days by oral administration daily with 80 mg/Kg of sunitinib malate (corresponding to 60 mg/Kg of the active ingredient) or vehicle solution, according to the manufacturer instructions. Ela-myc mice were treated when they were 2- or 4.5-months old, for incipient or advanced tumors, respectively; SCID Beige mice were treated when tumors reached 0.5 cm2.
Ela-myc mice were sacrificed when tumors compromised animal well-being, as judged by the protocol of Morton and Griffiths [63]. SCID Beige mice were treated with sunitinib or the control vehicle for 15 days and tumor size in all mice was measured weekly with a caliper. Mice were sacrificed, tumors were weighed, fixed in buffered formalin for 24 h, dehydrated and embedded in paraffin.
Histopathology and immunohistochemistry
For histopathological analysis, tumor sections from mice treated with sunitinib or control vehicle were stained with H&E and evaluated by two expert pathologists (M.I. and J.M-C.) to determine acinar ductal differentiation and the necrotic index [30, 62].
For immunohistochemistry, 5-μm sections from formalin-fixed paraffin-embedded tissue blocks were deparaffined and boiled with 0.01 M citrate buffer (pH 6.0) at 120°C for 10 min in a pressure cooker. Endogenous peroxidase activity was quenched with 3% H2O2, and samples were blocked in PBS with 1% BSA. Primary antibodies were added overnight at 4°C. Antibodies used for specific tissue immunostaining included anti-VEGFR2 (Cell Signaling Technology), anti-PDGFR-α (R&D) anti-PDGFR-β (Cell Signaling Technology), anti-vWF (Neomarkers), anti-cleaved caspase 3 (R&D systems) anti-P-Histone H3 (Ser10) (Millipore) and anti-Ki67 (Novo Castra). Negative controls were performed with pre-immune rabbit serum. As secondary antibodies, peroxidase-conjugated (Envision+, Dako) anti-rabbit Ig reagents were used. Reactions were developed using 3,3′-diaminobenzidine (DAB) as chromogenic substrate (Dako). Sections were counterstained with hematoxylin, dehydrated and mounted. An Olympus BX61 microscope was used for visualization, and images were acquired using CellSens software.
For quantification, 10 images were acquired at 10× for each tumor and analyzed with ImageJ software. To quantify angiogenesis and apoptosis, the area stained positive for vWF or active caspase 3, as indicated, was related to total area. Due to intrinsic differences in their proliferation, ductal and acinar cells were immunostained with Ki67 or P-Histone H3, respectively, as previously described [62]. For quantification, the percentage of Ki67 (in acinar regions) or P-Histone H3 positive cells (in ductal regions) were obtained by relating the area corresponding of nuclei positive for Ki67 or P-Histone H3 to the total nuclei area (positive for hematoxylin).
Cell lines
Emyc-1 and Emyc-10 cell lines were obtained from Ela-myc tumors as previously described [64, 65]. Cells were cultured in DMEM supplemented with 10% FBS at 5% CO2 and 37°C.
Western blots
Whole cell extracts were prepared with Laemmli buffer, and samples were boiled at 95°C for 5 min. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose filters for Western blot analysis. The primary antibodies used were: anti-VEGFR2 (Cell Signaling Technology), anti-PDGFR-α and anti-PDGFR-β (Santa Cruz), anti-cleaved caspase 3 (Cell Signaling Technology), anti-P-Histone H3 (Ser10) (Millipore) and anti-tubulin (Sigma). Specie-specific secondary antibodies conjugated to HRP (Dako) and ECL detection (Amersham) were used for band visualization.
Cell viability
In vitro cell viability of Emyc-1 cells was assessed with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide staining (MTT). Cells were seeded in 96-well plates in quintuplicate at a density of 1000 cells per well and grown in 2% FBS with the sunitinib vehicle (DMSO) or in the presence of sunitinib in increasing concentrations (at 1, 2, or 4 μM). Cell viability was measured daily by incubating cells with MTT (1 mg/mL) for 4h. The formazan precipitate was extracted with DMSO:isopropanol (1:4), and absorbance was taken at 570 nm on a multiwell-plate reader.
Immunofluorescence to detect apoptosis and proliferation
Cells seeded over sterile coverslips and grown in 10% FBS were fixed in 4% PFA. Cells were permeabilized with 0.2% Triton in PBS and blocked with 5% BSA, 0.1% Tween-20. Coverslips were incubated overnight with rabbit anti-P-Histone H3 (Ser10) (Millipore) to determine proliferation or with rabbit anti-cleaved caspase 3 (R&D) to determine apoptosis. Negative controls were incubated with an irrelevant rabbit IgG (data not shown). An anti-rabbit Alexa Fluor 488 (Invitrogen) was used as the secondary antibody. Coverslips were mounted with DAPI Fluoromont-G (Southern Biotech), and IF was detected with an Olympus BX61 Microscope. Ten fields were quantified per coverslip at 10×. The number of P-Histone H3 positive cells was manually counted and compared to the total number of cells (DAPI positive) for each experiment, which was quantified using an ImageJ macro developed in the CRG Advanced Light Microscopy Unit facility (CRG, Barcelona).
Statistical analysis
Statistical analyses were performed with SPSS version 12.0. Statistical significance cut-off was considered as p < 0.05. Kaplan-Meier analyses were used to establish survival curves, and comparisons were made using the log-rank test. The Student’s t-test was used with normally distributed data (in vitro) and the Mann-Whitney test was used in other occasions (in vivo).
ACKNOWLEDGMENTS
The authors would like to thank J.M. Caballero (PRBB Animal Facility, Barcelona) for help in sunitinib administration and F.X. Real (CNIO, Madrid) and J. Luna (IDIBAPS, Barcelona) for help with Ela-myc mice. We are very grateful to R. Peña and A.G. de Herreros (IMIM, Barcelona) for providing critical reagents, X. Sanjuán (CRG Advanced Light Microscopy Unit, Barcelona) for editing software for image analysis, A. Martñnez from the Medical Oncology Department (Hospital del Mar, Barcelona) for clinical advice, G. Gil for critical reading of the manuscript, L. Fàbregas for technical assistance, A. Flotats from Science&Design for help in graphic design and V. Raker for English proofreading and editing.
CONFLICTS OF INTEREST
The authors do not have any conflicts of interest.
GRANT SUPPORT
Supported by Spanish Ministerio de Economña y Competitividad/ ISCIII-FEDER (PI14/00125), RETIC Cancer RD12/0036/0051/FEDER, and the “Generalitat de Catalunya” (2014/SGR/143) to P.N., and Spanish Ministerio de Economña y Competitividad/ISCIII-FEDER (BIO2014-57716-C2-2-R, IIS10/00014) to C.F.
REFERENCES
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74:2913-2921.
2. Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol. 2015; 12:319-334.
3. La RS, Sessa F, Capella C. Acinar Cell Carcinoma of the Pancreas: Overview of Clinicopathologic Features and Insights into the Molecular Pathology. Front Med (Lausanne). 2015; 2:41.
4. Ito T, Igarashi H, Jensen RT. Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): recent insights and advances. J Gastroenterol. 2012; 47:941-960.
5. Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003; 9:327-337.
6. Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003; 2:471-478.
7. O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003; 101:3597-3605.
8. Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007; 25:884-896.
9. Seandel M, Shia J, Linkov I, Maki RG, Antonescu CR, Dupont J. The activity of sunitinib against gastrointestinal stromal tumor seems to be distinct from its antiangiogenic effects. Clin Cancer Res. 2006; 12:6203-6204.
10. Awasthi N, Schwarz MA, Schwarz RE. Antitumour activity of sunitinib in combination with gemcitabine in experimental pancreatic cancer. HPB (Oxford). 2011; 13:597-604.
11. Blumenthal GM, Cortazar P, Zhang JJ, Tang S, Sridhara R, Murgo A, Justice R, Pazdur R. FDA approval summary: sunitinib for the treatment of progressive well-differentiated locally advanced or metastatic pancreatic neuroendocrine tumors. Oncologist. 2012; 17:1108-1113.
12. Vinik AI, Raymond E. Pancreatic neuroendocrine tumors: approach to treatment with focus on sunitinib. Therap Adv Gastroenterol. 2013; 6:396-411.
13. Bergmann L, Maute L, Heil G, Russel J, Weidmann E, Koberle D, Fuxius S, Weigang-Kohler K, Aulitzky WE, Wormann B, Hartung G, Moritz B, Edler L, Burkholder I, Scheulen ME, Richly H. A prospective randomised phase-II trial with gemcitabine versus gemcitabine plus sunitinib in advanced pancreatic cancer: a study of the CESAR Central European Society for Anticancer Drug Research-EWIV. Eur J Cancer. 2015; 51:27-36.
14. O'Reilly EM, Niedzwiecki D, Hall M, Hollis D, Bekaii-Saab T, Pluard T, Douglas K, Abou-Alfa GK, Kindler HL, Schilsky RL, Goldberg RM. A Cancer and Leukemia Group B phase II study of sunitinib malate in patients with previously treated metastatic pancreatic adenocarcinoma (CALGB 80603). Oncologist. 2010; 15:1310-1319.
15. Yokoi K, Sasaki T, Bucana CD, Fan D, Baker CH, Kitadai Y, Kuwai T, Abbruzzese JL, Fidler IJ. Simultaneous inhibition of EGFR, VEGFR, and platelet-derived growth factor receptor signaling combined with gemcitabine produces therapy of human pancreatic carcinoma and prolongs survival in an orthotopic nude mouse model. Cancer Res. 2005; 65:10371-10380.
16. Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE. Evaluation of poly-mechanistic antiangiogenic combinations to enhance cytotoxic therapy response in pancreatic cancer. PLoS One. 2012; 7:e38477.
17. Awasthi N, Zhang C, Schwarz AM, Hinz S, Schwarz MA, Schwarz RE. Enhancement of nab-paclitaxel antitumor activity through addition of multitargeting antiangiogenic agents in experimental pancreatic cancer. Mol Cancer Ther. 2014; 13:1032-1043.
18. Tran Cao HS, Bouvet M, Kaushal S, Keleman A, Romney E, Kim G, Fruehauf J, Imagawa DK, Hoffman RM, Katz MH. Metronomic gemcitabine in combination with sunitinib inhibits multisite metastasis and increases survival in an orthotopic model of pancreatic cancer. Mol Cancer Ther. 2010; 9:2068-2078.
19. Casneuf VF, Demetter P, Boterberg T, Delrue L, Peeters M, Van DN. Antiangiogenic versus cytotoxic therapeutic approaches in a mouse model of pancreatic cancer: an experimental study with a multitarget tyrosine kinase inhibitor (sunitinib), gemcitabine and radiotherapy. Oncol Rep. 2009; 22:105-113.
20. Blansfield JA, Caragacianu D, Alexander HR, III, Tangrea MA, Morita SY, Lorang D, Schafer P, Muller G, Stirling D, Royal RE, Libutti SK. Combining agents that target the tumor microenvironment improves the efficacy of anticancer therapy. Clin Cancer Res. 2008; 14:270-280.
21. Olson P, Chu GC, Perry SR, Nolan-Stevaux O, Hanahan D. Imaging guided trials of the angiogenesis inhibitor sunitinib in mouse models predict efficacy in pancreatic neuroendocrine but not ductal carcinoma. Proc Natl Acad Sci U S A. 2011; 108:E1275-E1284.
22. Sandgren EP, Quaife CJ, Paulovich AG, Palmiter RD, Brinster RL. Pancreatic tumor pathogenesis reflects the causative genetic lesion. Proc Natl Acad Sci U S A. 1991; 88:93-97.
23. Grippo PJ, Sandgren EP. Acinar-to-ductal metaplasia accompanies c-myc-induced exocrine pancreatic cancer progression in transgenic rodents. Int J Cancer. 2012; 131:1243-1248.
24. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008; 3:157-188.
25. Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol. 2002; 15:462-469.
26. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008; 321:1801-1806.
27. Hessmann E, Schneider G, Ellenrieder V, Siveke JT. MYC in pancreatic cancer: novel mechanistic insights and their translation into therapeutic strategies. Oncogene. 2016; 35:1609-1618.
28. Skoudy A, Hernandez-Munoz I, Navarro P. Pancreatic Ductal Adenocarcinoma and Transcription Factors: Role of c-Myc. J Gastrointest Cancer. 2011; 42:76-84.
29. Ischenko I, Zhi J, Moll UM, Nemajerova A, Petrenko O. Direct reprogramming by oncogenic Ras and Myc. Proc Natl Acad Sci U S A. 2013; 110:3937-3942.
30. Martinez-Bosch N, Iglesias M, Munne-Collado J, Martinez-Caceres C, Moreno M, Guerra C, Yelamos J, Navarro P. Parp-1 genetic ablation in Ela-myc mice unveils novel roles for Parp-1 in pancreatic cancer. J Pathol. 2014; 234:214-227.
31. Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007; 6:1186-1197.
32. Burris HA, III, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997; 15:2403-2413.
33. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364:1817-1825.
34. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van CE, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013; 369:1691-1703.
35. Hayes AJ, Li LY, Lippman ME. Anti-vascular therapy: a new approach to cancer treatment. West J Med. 2000; 172:39-42.
36. Hotz HG, Gill PS, Masood R, Hotz B, Buhr HJ, Foitzik T, Hines OJ, Reber HA. Specific targeting of tumor vasculature by diphtheria toxin-vascular endothelial growth factor fusion protein reduces angiogenesis and growth of pancreatic cancer. J Gastrointest Surg. 2002; 6:159-166.
37. Buchler P, Reber HA, Ullrich A, Shiroiki M, Roth M, Buchler MW, Lavey RS, Friess H, Hines OJ. Pancreatic cancer growth is inhibited by blockade of VEGF-RII. Surgery. 2003; 134:772-782.
38. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009; 324:1457-1461.
39. Kindler HL, Ioka T, Richel DJ, Bennouna J, Letourneau R, Okusaka T, Funakoshi A, Furuse J, Park YS, Ohkawa S, Springett GM, Wasan HS, Trask PC, Bycott P, Ricart AD, Kim S, Van CE. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol. 2011; 12:256-262.
40. Cabebe E, Fisher GA. Clinical trials of VEGF receptor tyrosine kinase inhibitors in pancreatic cancer. Expert Opin Investig Drugs. 2007; 16:467-476.
41. Cardin DB, Goff L, Li CI, Shyr Y, Winkler C, DeVore R, Schlabach L, Holloway M, McClanahan P, Meyer K, Grigorieva J, Berlin J, Chan E. Phase II trial of sorafenib and erlotinib in advanced pancreatic cancer. Cancer Med. 2014; 3:572-579.
42. de BS, Herlin P, Christensen JG, Lemoisson E, Gauduchon P, Raymond E, Guillamo JS. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007; 9:412-423.
43. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003; 111:1287-1295.
44. Jaster R. Molecular regulation of pancreatic stellate cell function. Mol Cancer. 2004; 3:26.
45. Prenen H, Cools J, Mentens N, Folens C, Sciot R, Schoffski P, Van OA, Marynen P, Debiec-Rychter M. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006; 12:2622-2627.
46. Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, Armand JP, Scigalla P, Raymond E. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006; 24:25-35.
47. Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, Booth BP, Verbois SL, Morse DE, Liang CY, Chidambaram N, Jiang JX, Tang S, Mahjoob K, Justice R, Pazdur R. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007; 13:1367-1373.
48. Cuneo KC, Geng L, Fu A, Orton D, Hallahan DE, Chakravarthy AB. SU11248 (sunitinib) sensitizes pancreatic cancer to the cytotoxic effects of ionizing radiation. Int J Radiat Oncol Biol Phys. 2008; 71:873-879.
49. Passadouro M, Pedroso de Lima MC, Faneca H. MicroRNA modulation combined with sunitinib as a novel therapeutic strategy for pancreatic cancer. Int J Nanomedicine. 2014; 9:3203-3217.
50. Tuveson D, Hanahan D. Translational medicine: Cancer lessons from mice to humans. Nature. 2011; 471:316-317.
51. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012; 21:418-429.
52. Rucki AA, Zheng L. Pancreatic cancer stroma: understanding biology leads to new therapeutic strategies. World J Gastroenterol. 2014; 20:2237-2246.
53. Becher OJ, Holland EC. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006; 66:3355-3358.
54. Henderson VC, Demko N, Hakala A, MacKinnon N, Federico CA, Fergusson D, Kimmelman J. A meta-analysis of threats to valid clinical inference in preclinical research of sunitinib. Elife. 2015; 4: e08351.
55. Reni M, Cereda S, Milella M, Novarino A, Passardi A, Mambrini A, Di LG, Aprile G, Belli C, Danova M, Bergamo F, Franceschi E, Fugazza C, Ceraulo D, Villa E. Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: a phase II randomised trial. Eur J Cancer. 2013; 49:3609-3615.
56. Bharthuar A, Pearce L, Litwin A, LeVea C, Kuvshinoff B, Iyer R. Metastatic pancreatic adenocarcinoma and renal cell carcinoma treated with gemcitabine and sunitinib malate. A case report. JOP. 2009; 10:523-527.
57. Pietras K, Hanahan D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol. 2005; 23:939-952.
58. Kulke MH, Lenz HJ, Meropol NJ, Posey J, Ryan DP, Picus J, Bergsland E, Stuart K, Tye L, Huang X, Li JZ, Baum CM, Fuchs CS. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008; 26:3403-3410.
59. Wiedmann MW, Mossner J. Safety and efficacy of sunitinib in patients with unresectable pancreatic neuroendocrine tumors. Clin Med Insights Oncol. 2012; 6:381-393.
60. Lowery MA, Klimstra DS, Shia J, Yu KH, Allen PJ, Brennan MF, O'Reilly EM. Acinar cell carcinoma of the pancreas: new genetic and treatment insights into a rare malignancy. Oncologist. 2011; 16:1714-1720.
61. Wisnoski NC, Townsend CM, Jr., Nealon WH, Freeman JL, Riall TS. 672 patients with acinar cell carcinoma of the pancreas: a population-based comparison to pancreatic adenocarcinoma. Surgery. 2008; 144:141-148.
62. Martinez-Bosch N, Fernandez-Barrena MG, Moreno M, Ortiz-Zapater E, Munne-Collado J, Iglesias M, Andre S, Gabius HJ, Hwang RF, Poirier F, Navas C, Guerra C, Fernandez-Zapico ME, Navarro P. Galectin-1 Drives Pancreatic Carcinogenesis through Stroma Remodeling and Hedgehog Signaling Activation. Cancer Res. 2014; 74:3512-3524.
63. Morton DB, Griffiths PH. Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. Vet Rec. 1985; 116:431-436.
64. Biliran H, Jr., Wang Y, Banerjee S, Xu H, Heng H, Thakur A, Bollig A, Sarkar FH, Liao JD. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin Cancer Res. 2005; 11:6075-6086.
65. Jose A, Sobrevals L, Miguel Camacho-Sanchez J, Huch M, Andreu N, Ayuso E, Navarro P, Alemany R, Fillat C. Intraductal delivery of adenoviruses targets pancreatic tumors in transgenic Ela-myc mice and orthotopic xenografts. Oncotarget. 2013; 4:94-105. doi: 10.18632/oncotarget.795.