
INTRODUCTION
Few occupational or environmental hazards are unambiguously linked to the development of myeloid neoplasms. This is partly due to the uncertainty between the time of exposure and the appearance of symptoms. In addition, criteria for diagnosing these diseases have dramatically changed over the years, in particular for myelodysplastic syndrome (MDS), which complicates the evaluation of old patient records. Nevertheless, decades of follow-up studies and reexamining pathology reports show that adults who were exposed at the work place to cumulative high levels, or chronic low levels of benzene have an increased risk of developing MDS or acute myeloid leukemia (AML), respectively [1–3].
Benzene is a colorless volatile liquid hydrocarbon found in coal tar and petroleum and is used to make numerous chemical products including detergents, insecticides and motor fuels [4, 5]. The primary benzene metabolite to cause genomic damage is 1,4-benzoquinone (BQ) and is believed to be responsible for the myelotoxicity/myeloid neoplasms observed in the bone marrow of people who have been exposed to increased levels of benzene [4]. In addition to specific industry-related work places, benzene concentrations are also significantly higher in and around cities with a high coal or oil-based energy consumption, as well as in rural areas subject to heavy pesticide use. This raises the question whether environmental benzene pollution can be a contributing factor to MDS and AML development. In support of this notion is the increased incidence of childhood leukemia observed in Harris county, Texas (Houston area), which houses several petroleum and chemical industries that are associated with air pollutants, including benzene [6]. In addition, several studies report that MDS is diagnosed on average a decade earlier in Asian countries compared to Western countries [7–11]. Since the former countries use mostly coal to fuel their industries [12] and households, systemic exposure to environmental toxins such as benzene, could be responsible for their earlier onset of MDS.
An alternative explanation for the difference in age of MDS onset between Western and Asian populations is a difference in genetic make-up. There is no doubt that an individual’s machinery responsible for proper genome maintenance suppresses hematologic cancers [13]. This is particularly apparent in diseases characterized by poor DNA repair capacity [14, 15]. For example, patients with Fanconi Anemia (FA) are defective for genome maintenance and exhibit a high incidence of MDS and AML [16]. Moreover, alternative SNPs in multiple DNA repair genes were found to associate with hematotoxicity in adults routinely exposed to benzene [17]. The difference in childhood leukemia onset in specific Texas counties may also have an underlying genetic basis. Texas is home to a large Hispanic population and several studies have shown that Hispanic children exhibit a significantly higher incidence and worse outcome of acute lymphocytic leukemia (ALL) compared to non-Hispanic children. Specifically, Hispanic children in Texas who developed ALL overrepresented several polymorphisms in genes known to associate with cancer development when mutated [18, 19]. This same group of children exhibited a significantly higher risk for developing secondary MDS/AML after receiving etoposide (a topoisomerase type II inhibitor that was part of their ALL treatment) [20]. Thus, occupational and environmental exposures to benzene, as well as poor DNA damage response/repair can enhance the risk for the development of hematologic cancers like MDS and AML.
Very little is known about the mechanisms required to cope with benzene-induced DNA damage. This lack of understanding hampers a more detailed assessment of the risk benzene exposure poses to people and our ability to identify those at high risk for MDS and AML. The current study was initiated to increase our understanding of the consequences of benzene-induced DNA damage and the mechanisms required to repair it. We found that the benzene metabolite, BQ induced chromosomal breaks and rearrangements as well as stalled replication forks, which required DSB repair and the FA pathway to correct. Furthermore, BQ directly interfered with the ability of type 1 topoisomerase (topo 1) to nick DNA and relieve supercoiling. Topo 1 interference is consistent with the observations that BQ-induced damage causes replication fork regression that could lead to chromosomal breaks and rearrangements, especially if DSB repair and FA pathways are compromised. Thus, these data support the observations that benzene enhances risk of MDS and AML especially for those with compromised genome maintenance capacity.
RESULTS
Cells defective for DSB repair and replication fork stability are hypersensitive to BQ
We previously developed a screening system to identify the DNA repair pathway(s) most important for repairing DNA lesions induced by a given genotoxin [21]. This screen will generate a genotoxic profile of the toxin under investigation and it is based on a comprehensive set of mouse embryonic stem (ES) cells defective for specific DNA repair pathways, including those that repair base lesions, replication errors, double strand breaks (DSBs) and interstrand and intrastrand crosslinks (Table 1) [21]. To create a BQ genotoxic profile, we performed a dose response curve for each mutant cell line to determine the threshold BQ concentrations that reduce cell survival [22]. Mutant cells that are more sensitive to threshold BQ concentrations as compared to their parental control reveal a pathway important for correcting BQ-induced damage. Thus, this screening method takes an unbiased approach to discover the DNA repair pathways most important for correcting BQ-induced damage.
Table 1: Summary of mutant ES cells
Control cells |
Gene |
Mutations |
Function |
|---|---|---|---|
AB1.1 |
Msh2 |
-/- |
MMR |
AB2.2 |
Brca2 |
Exon 27 deletion |
HR |
Blm |
88% decrease |
Helicase/HR |
|
Recql5 |
-/- |
Helicase/HR |
|
Trex2 |
-/- |
Exonuclease/RF |
|
FancB |
Exon 2 deletion |
ICLR/RF |
|
B44 |
Xpa |
-/- |
NER |
Xpc |
-/- |
NER |
|
J1 |
Ku70 |
-/- |
cNHEJ |
TC1 |
H2AX |
-/- |
DDR/HR |
Brca1 |
BRCT deletion |
DDR/HR/NHEJ |
|
IB10 |
Rad18 |
-/- |
Lesion bypass |
E14 (IB10) |
Rad52 |
-/- |
HR |
Rad54 |
-/- |
HR |
|
Mus81 |
-/- |
Endonuclease/HR |
|
Ercc1 |
-/- |
NER/HR/ICLP |
Cells were used that were ablated for nucleotide excision repair (NER) (Xpa [73], Xpc [74]), mismatch repair (MMR, Msh2) [75], lesion bypass (Rad18) [76], the Fanconi anemia (FA) pathway (Fancb) [77], nonhomologous end joining (NHEJ, Ku70) [78]. Complete ablation of homologous recombination (HR) is cell lethal [79]; therefore, null cells were used for genes that contribute to, but are not essential for HR (H2ax [80], Rad52 [81], Rad54 [82]). Cells were used that are partially defective for essential proteins that include a deletion of Brca2 exon 27 [26] and deletion of Brca1 exon 11 [83]. Cells were used that are defective for HR regulation that include mutations in the helicases Blm [84] and Recql5 [85]. Cells were used that are defective for endonucleases (Mus81 [86] and Ercc1 [87]) that can be used during HR and interstrand crosslink repair (ICLR) and exonucleases (Trex2) [88] that can be used for lesion bypass.
The mutant cells used for the BQ genotoxic profile are summarized in Table 1. The survival difference between mutant relative to control cells is shown at BQ concentrations that reduce mutant cell survival by 90% and 99% (black and grey bars, respectively). For example, at a BQ dose that reduces the survival of Brca1-mutant cells by 90% and 99%; the mutant cells exhibited an 8-fold and a 17-fold increase in sensitivity (measured as reduced cell survival) compared to control cells, respectively. This observation suggests BQ induces DNA breaks and destabilizes replication forks since BRCA1 is needed to address these problems as a member of HR. In support, cells mutated for other DSB repair and replication fork maintenance genes also exhibited >5-fold hypersensitivity to BQ (Figure 1A). These include cells defective for FA (Fancb), nonhomologous end joining (NHEJ, Ku70) and interstrand crosslink repair (ICLR)/HR (Ercc1). By comparison, cells with a mutation in a lesion bypass gene, Trex2, caused BQ-resistance supporting the notion that DSB repair and replication fork maintenance are important for correcting BQ-induced lesions since Trex2-deletion is known to increase HR and NHEJ [23, 24]. The Brca2-mutant cells did not exhibit profound hypersensitivity even though BRCA2 is important for DSB repair and replication fork maintenance [25]. However, these cells produce wild type levels of a C-terminally truncated protein that is defective in RAD51 filament stability which causes only a minor phenotype [25–28]. Taken together, these data suggest that DSB repair and replication fork maintenance are essential for efficiently repairing BQ-induced damage.
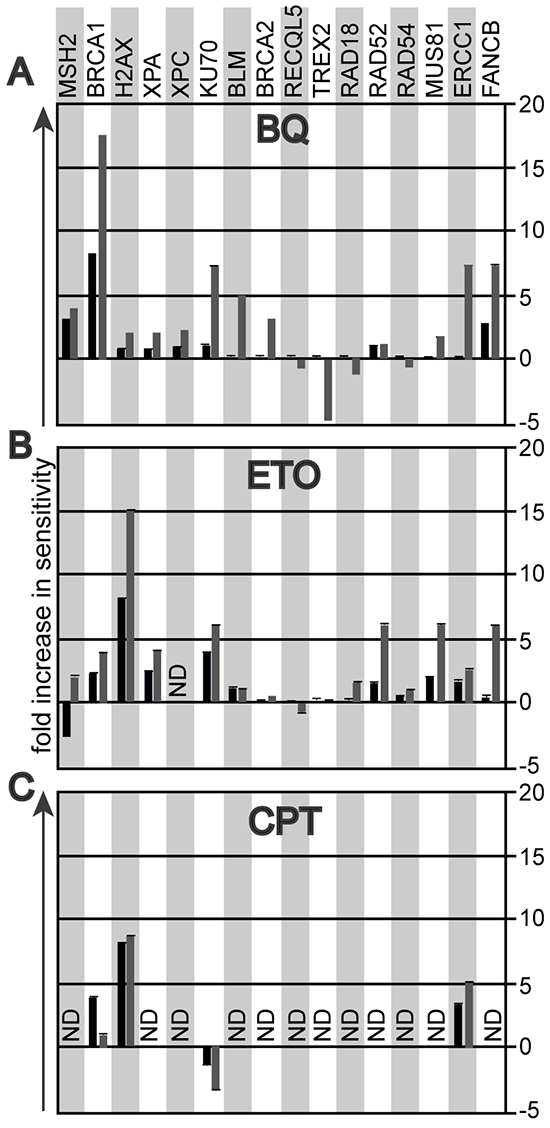
Figure 1: The genotoxic profile that compares the survival fraction of mutant ES cells to their parental controls at 10% (black bar) and at 1% (grey bar) cell survival. A. Exposure to 1,4-Benzoquinone (BQ). B. Exposure to etoposide (ETO). C. Exposure to camptothecin (CPT).
Others have shown that BQ inhibits type II topoisomerases [29], leading to the formation of apoptosis-inducing type I topoisomerase cleavage complexes [30]. We therefore hypothesized that the genotoxic profile of BQ could resemble that of a type II topoisomerase poison such as etoposide (ETO) and potentially that of a type 1 topoisomerase inhibitor such as camptothecin (CPT). Indeed, like BQ, ETO exposure caused hypersensitivity in cells defective in DSB repair and replication fork maintenance (Figure 1B). Curiously, Msh2-/- cells exposed to ETO show antithetical responses depending on dose. It is possible at the lower dose Msh2-/- cells were resistant because MSH2 hindered replication fork progression while at the higher dose MSH2 were hypersensitive because MSH2 was needed to corrected damage that severely disabled replication or induced apoptosis. We then assessed the dose response to CPT in the mutant ES cell lines most sensitive to BQ and ETO (cells mutated for Brca1, H2ax, Ku70 and Ercc1). Cells defective for HR (Brca1, H2ax) and ICLR (Ercc1) were hypersensitive to CPT, similar to BQ and ETO. Yet the NHEJ-defective cells (Ku70) were mildly resistant to CPT, suggesting that NHEJ performs a toxic function when encountering CPT-induced damage, unlike the response to BQ- and ETO-induced lesions. Thus, DSB repair and replication fork maintenance appear to be critical for correcting lesions caused by BQ, ETO and CPT.
BQ causes less chromosomal damage than ETO and CPT
The BQ genotoxic profile predicts that cells defective in DSB repair and replication fork maintenance will exhibit extensive chromosomal damage after BQ exposure; therefore, we evaluated chromosome integrity of relevant ES cell lines using two-color fluorescence in situ hybridization (FISH) on metaphase spreads (MPSs). DAPI counterstained chromosomes were stained with a telomere and a pericentromere probe [31]. We scored isochromatid breaks, chromatid breaks, radials and EPTs (extra pericentromeres and telomeres) (Figure 2A) [32]. Chromatid breaks indicate one-ended breaks observed at collapsed replication forks while isochromatid breaks indicate failed strand exchange intermediates that break both chromatids. Radials suggest chromosomal structures that result from the fusion of multiple chromosomes, such as those commonly observed in cells derived from FA patients after exposure to DNA crosslinking agents [33]. EPTs are complex rearrangements that occur in HR-defective cells that implicate multiple fusions possibly due to faulty replication or the imprecise joining of multiple DSBs [32]. Thus, two-color FISH will detect a range of chromosomal defects.
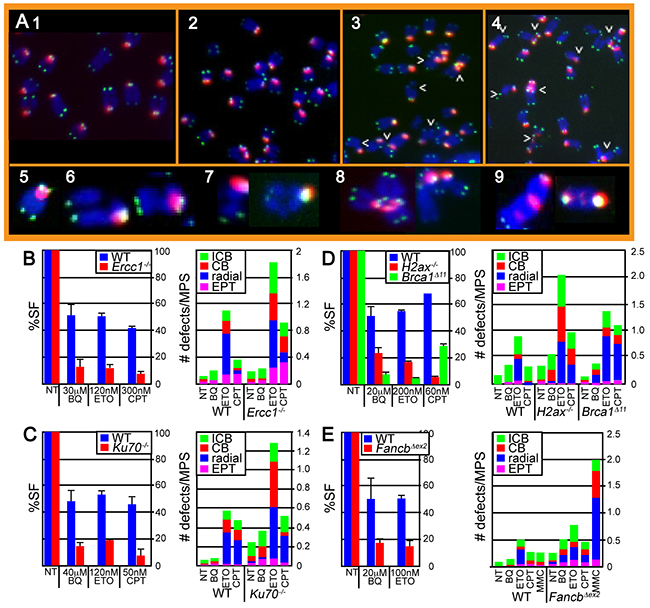
Figure 2: Evaluation of chromosome damage in metaphase spreads (MPS) after ES cells were exposed to BQ, ETO and CPT. A. Images of H2ax-/- cells exposed to 1) nothing, 2) BQ, 3) CPT and 4) ETO. Arrowheads point to chromosomal abnormalities. Enlarged representative examples of chromosomes include 5) normal, 6) isochromatid break (ICB), 7) chromatid break (CB), 8) radial and 9) extrapericentromeres and telomeres (EPT). B–E. The survival fraction (%SF) is shown on the left panel and the # of chromosomal defects is shown on the right panel. (B) Wild type and Ercc1-mutant IB10 cells. (C) Wild type and Ku70-mutant J1 cells. (D) Wild type and H2ax- and Brca1-mutated TC1 cells. (E) Wild type and Fancb-mutated AB2.2 cells. Cells were also exposed to an equivalently toxic dose of Mitomycin C (MMC), a crosslinking agent that is known to be very toxic to FA-defective cells. The concentration for CPT (100 nM, 16 hours) and MMC (30 nM, 16 hours) results in ~ 10% and 90% survival for control cells and ~ 10% and <0.001% survival for FancbΔex2 cells as previously reported [38]. Note that MMC induces a much larger level of cell death and radials relative to control cells than the other agents relative to control cells demonstrating that FancbΔex2 cells are particularly susceptible to MMC as compared to the other agents. The total number of MPS observed for each bar and statistics are shown in supplemental tables 1 and 2, respectively.
BQ-induced damage was compared to that of ETO and CPT because all three genotoxins showed similar results in the screening assay. We focused on those mutant ES cells that showed a hypersensitive phenotype in response to BQ (Ercc1-/-, Ku70-/-, H2ax-/-, Brca1Δ11/Δ11 and FancbΔex2 ES cells). Physiologically comparable doses of genotoxins were used that produced a survival fraction of ~40-70% and ~5-20% for control and mutant cells, respectively (Figure 2B–2E, left panels). Therefore, these genotoxins are being compared at comparably toxic doses. At these doses, BQ caused fewer chromosomal defects than either ETO or CPT (Figure 2B–2E, right panels, Supplementary Table S1A-S1D, Supplementary Table S2A-S2D). Yet, this difference dissipates in FancbΔex2 cells implicating the FA pathway as central for repairing BQ-induced damage. FANCB is an essential member of the FA core complex [34] that is capable of monoubiquitinating FANCD2 in a minimal subcomplex [35] and its disruption completely destroys core complex function [36]. The FA pathway is important for replication fork maintenance, in particular protection of the nascent strand [37, 38]. These results suggest that BQ-mediated DNA damage has the potential to disrupt replication.
BQ is more efficient at stalling replication forks than ETO and CPT
DNA fiber analysis was used to measure replication fork restart in response to BQ in control and FancbΔex2 cells since the FA pathway is important for coping with BQ-induced DNA damage and since FA stabilizes replication forks [37, 38]. In this assay, BQ was compared to a positive control, hydroxyurea (HU). HU inhibits ribonucleotide reductase, which depletes nucleotides [39] to impair the restart of replication forks [25]. Physiologically comparable doses of genotoxins (1.5 hr of 10 μM BQ or 0.5 mM HU) were used that produced a survival fraction of ~80-90% in control cells (Figure 3A). This particular dose of HU does not cause breaks [23, 25] and has a mild effect on replication fork restart and origin firing in control cells (Figure 3B, Supplementary Tables S3 & S4). In contrast, BQ significantly reduced levels of restart while it increased levels of new origin firing (Figure 3B). In FancbΔex2 cells, both BQ and HU reduced restart and new origins, with BQ being more severe (Figure 3C). A higher BQ concentration did not exacerbate these observations, suggesting the lower dose already produced the maximal effect (Figure 3B, 3C).
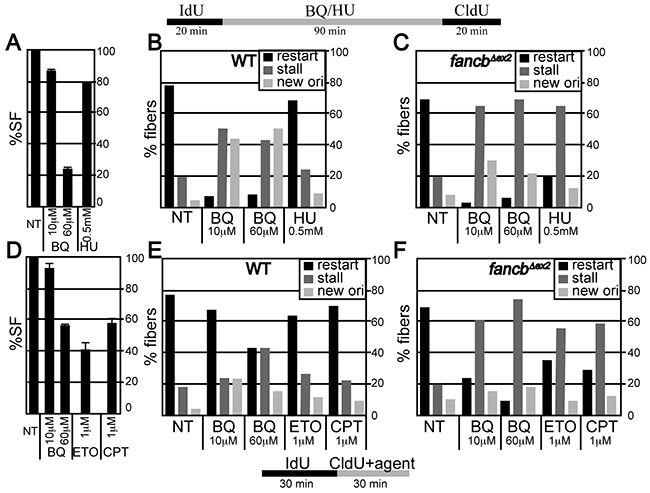
Figure 3: Fiber analysis. A-C. ES cells were exposed to IdU for 20 minutes and then agent (BQ or HU) for 90 minutes and then CldU for 20 minutes. (A) Percent survival fraction (%SF) using the identical condition as the fiber analysis. (B) Fiber analysis in wild type AB2.2 cells. (C) Fiber analysis in Fancb-mutant cells. D-E. ES cells were exposed to IdU for 30 minutes and CldU + agent for 30 minutes. (D) Percent survival fraction (%SF) using the identical conditions for the fiber analysis. E. Fiber analysis in wild type AB2.2 cells. F. Fiber analysis in Fancb-mutant cells. The total number of fibers observed for each bar and statistics are shown in supplemental tables S3 and S4, respectively.
BQ’s impact on replication fork restart was compared to that of ETO and CPT, in control and FancbΔex2 cells. Physiologically comparable doses of genotoxin (0.5 hr of 60 μM BQ or 1 μM ETO/CPT) were used, producing a survival fraction of ~40-60 % for control cells (Figure 3D). In control cells, BQ reduced replication fork restart more than ETO and CPT (Figure 3E). By contrast in FancbΔex2 cells, all three genotoxins reduced replication fork restart with BQ being the most severe (Figure 3F). Thus, BQ blocks replication fork restart more than HU, ETO and CPT, which is exacerbated in FA-defective cells.
BQ is not efficient at inducing ATR/DNA-PKCS responses to stalled replication forks
A fork that fails to restart can be temporarily stalled or more severely collapsed, possibly without a replisome [40]. The latter is more likely to form an intermediate structure amenable to a chromosomal rearrangement with potential for disease development, yet they are indistinguishable at the level of fiber analysis. Therefore, iPOND was used to observe the severity of the defect in replication fork restart by observing the phosphorylation pattern of RPA 32 [32, 41]. RPA 32 associates with single strand DNA at replication forks [42]. ATR phosphorylates serine 33 for a mild response while DNA-PKCS phosphorylates serines 4 and 8 for a severe response [43]. For ES cells, a low HU dose (0.5 mM, 1.5 hours) induces an ATR response while a high HU dose (4 mM, 5 hours) induces both an ATR and a DNA-PKCS response [25]. Previously we showed that the high HU dose produced more chromatid breaks; thus indicating collapsed forks with DSBs [25]. We compared similar physiologically toxic doses of BQ, ETO and CPT to that of low and high dose HU (Figure 4A). We found that all genotoxins caused an ATR response at the high doses but only ETO and HU caused this response at the low doses (Figure 4B, 4C). Furthermore, only high dose HU caused a DNA-PKCS response (Figure 4B, 4D). For confirmation, we also purified γH2AX since it recognizes DNA DSBs [44] and single strand DNA at stalled replication forks [41]. We found that γH2AX levels directly correlated with the severity of the response such that the highest levels purified in HU exposed cells followed by ETO, CPT and BQ. Thus, the purification of γH2AX correlates with dose severity similar to RPA 32 phosphorylation. Therefore, from the genotoxins tested here, BQ is efficient at inhibiting fork restart (fiber analysis), but not efficient at inducing an ATR/DNA-PKCS response or a γH2AX response (iPOND).
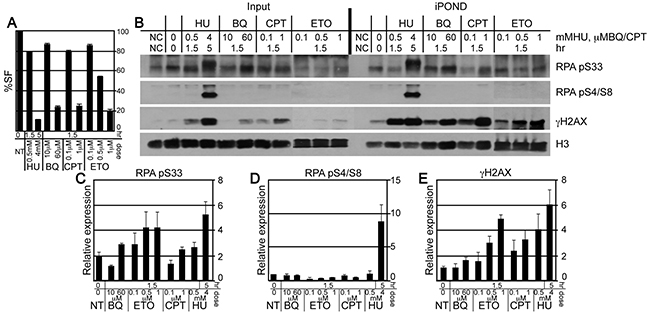
Figure 4: The purification of γH2AX and RPA at the nascent replication strand using iPOND. A. The percent survival fraction (%SF) using the same condition as for iPOND. B. Western blot to evaluate the protein concentrations at purified nascent replication strands. C-E. Graphs that depict the quantitation of 3 Western blots for (C) γH2AX, (D) RPA pS33 and (E) RPA pS4/S8. Error bars are shown for the average of 3 experiments.
BQ causes stalled replication forks to regress
We next observed the nature of individual stalled forks and DSBs induced by BQ since fibers suggest BQ efficiently inhibits fork restart but iPOND suggests minimal ATR/DNA-PKCS responses indicating that BQ-induced stalled forks are stabilized and not subject to these responses. Previously, CPT was shown to induce stalled forks that regress and form a chicken foot (Figure 5A). Fork regression could stabilize stalled forks to minimize ATR/DNA-PKCS responses. The localization of γH2AX and 53BP1 foci can be used to identify regressed forks. Similar to γH2AX, 53BP1 associates with damaged DNA to form nuclear foci [45]. Nuclei without foci implicate little to no damage (Figure 5B, 1st row). Nuclei with colocalized foci (a merge of γH2AX and 53BP1) implicate replication-independent damage that does not cause chicken feet as seen after exposure to γ-radiation (Figure 5B, 2nd row) [46]. By contrast nuclei with single-protein foci (either γH2AX or 53BP1) implicate replication-dependent damage that cause chicken feet as seen after exposure to CPT (Figure 5B, 3rd and 4th rows). Furthermore, PARP1 stabilizes chicken feet by inhibiting RECQ1 helicase and the PARP1 inhibitor, olaparib (OLA) reduces chicken feet [47, 48] such that exposure to OLA will reduce the number of nuclei with single-protein foci and an increase of nuclei with merged foci [47, 48]. Thus, the analysis of γH2AX and 53BP1 foci will measure the presence of PARP1-stabilized chicken feet.
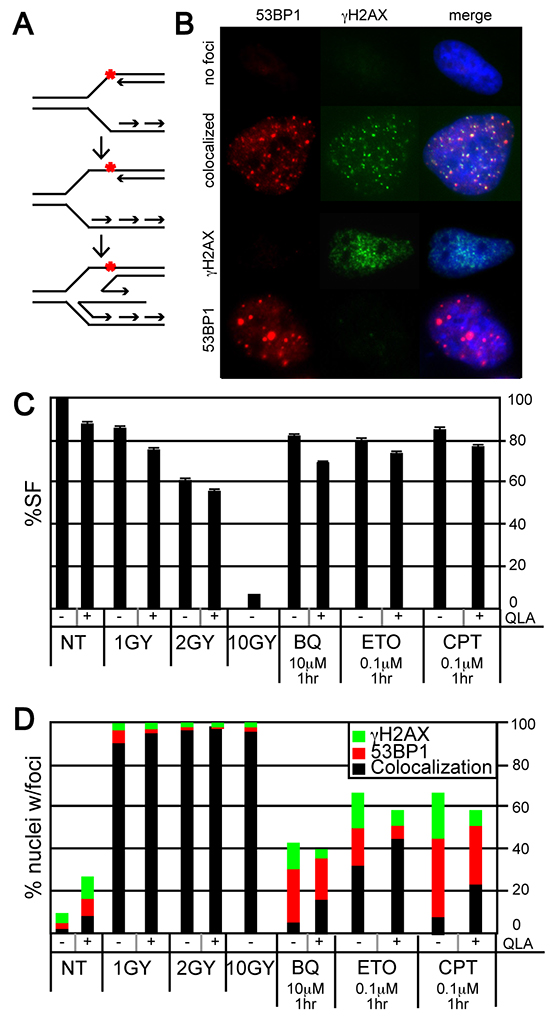
Figure 5: Evaluation of γH2AX and 53BP1 foci in HeLa cells exposed to BQ. A. The formation of a regressed fork (chicken foot). The red asterix is a symbol for DNA damage that stalls a fork. This damage could be a CPT-type 1 topoisomerase cleavage complex. B. Representative examples of nuclei with no foci, colocalized foci, γH2AX foci, and 53BP1 foci. C. Survival fraction after exposure to ionizing γ-radiation [IR: 1-10 Gray (Gy)], olaparib (OLA, 10 μM), BQ, ETO and CPT. D. The percentage of nuclei with separated or colocalized γH2AX and 53BP1 foci. Ten or more foci are needed to be positive. The total number of nuclei observed for each bar and statistics are shown in supplementary tables S5 and S6, respectively.
We evaluated γH2AX and 53BP1 foci to determine if BQ induced PAPR1-stabilized chicken feet. Gamma-radiation and CPT were used as controls since both cause DSBs but only CPT promotes chicken feet. Cells were exposed to a variety of γ-radiation doses that result in a survival fraction of ~97% (1Gy), ~60% (2 Gy) and ~7% (10 Gy). A dose of genotoxin that is physiologically comparable to the milder γ-radiation doses was used for the other genotoxins (Figure 5C). In addition, a mild OLA dose was used that had only minimal or no impact on cell survival in the presence of genotoxin (Figure 5C). As expected, γ-radiation (with or without OLA) resulted in a strong majority of nuclei with colocalized foci (no chicken feet) while CPT resulted in a majority of nuclei with single-protein foci (chicken feet) and OLA reduced the proportion of these nuclei (implicating PARP1-stabilized regressed forks) (Figure 5D, Supplementary Tables S5 & S6). Exposure to BQ was almost identical to CPT (except BQ-exposed cells had fewer nuclei with foci) while exposure to ETO was intermediate to γ-radiation and CPT. Thus, BQ appears to cause PARP1-stabilized chicken feet much like CPT, suggesting a similar mechanism of action to this type 1 topoisomerase inhibitor.
BQ inhibits the function of topoisomerase 1
The γH2AX and 53BP1 foci analysis supports the possibility that BQ directly inhibits type 1 topoisomerases. To test this notion, we used a standard biochemical assay that measures nicking and relaxing of a supercoiled DNA substrate. ETO (100 μM), a type 2 topoisomerase inhibitor, served as a negative control and did not nick or relax the supercoiled substrate, while CPT (500 μM) served as a positive control and indeed inhibited the relaxation of nicked circular DNA (Figure 6). Similar to CPT, BQ progressively inhibited the relaxing of nicked circular DNA from 20-70 μM and progressively inhibited the nicking of supercoiled DNA from 70-300 μM. Thus, BQ directly interferes with topoisomerase I activity.

Figure 6: BQ inhibits type 1 topoisomerase (topo 1). CPT is a positive control and ETO is a negative control. The relaxed DNA shown in lane 19 is a control that came with the kit.
DISCUSSION
Here we explore the nature of BQ genotoxicity since it is the primary metabolite suspected to cause the hematopoietic damage observed in people exposed to benzene. A non-biased approach was taken in mouse ES cells to identify the most critical pathways that address BQ-induced DNA damage. We found that DSB repair and replication fork maintenance pathways were essential for addressing these lesions. Moreover, we discovered that BQ interfered with type 1 topoisomerase which is consistent with a pathway necessary to maintain cell survival, replication fork stability and genome integrity.
For this proposal we performed our screen in mouse ES cells and comparisons to other cell types should be made with an understanding of their differences and similarities. One difference from many cells is that p53 exhibits some, but not all, its functions. Specifically, ES cells do not exhibit a p53/p21-mediated G1/S checkpoint even though they exhibit certain hallmarks like an IR-induced ATM/ATR response and p53-mediated increase in p21 transcription. In spite of these characteristics, there is no increase in p21 protein due to epigenetic regulation and proteasome-mediated degradation [49]. This is likely to prevent differentiation [50]. However, this p53-mediated response does not seem to be important for suppressing cancer since mice defective for it, but not other p53 responses (p533KR/3KR) [51] and mice deleted for p53 DNA damage targets (p21-/-, Puma-/-, Noxa-/-) [52] do not exhibit early lymphomas and sarcomas as do p53-null mice [53]. In addition, there are intra S- and G2 checkpoints that are independent of p53 [54]. Moreover, human ES cells commit to apoptosis instead of checkpoint activation when exposed to DNA replication inhibitors [55] and our data concurs for mouse ES cells [38]. These qualities should be understood when using mouse ES cells in order to fairly compare these cells to other cell types like cancer cells and hematopoietic stem cells (HSCs).
There are similarities between mouse ES cells to cancer cells and HSCs. Like ES cells, cancer cells are often mutant for p53 (hence no G1/S checkpoint) [56] with elevated glycolysis (Warburg effect) [57–60]. They both are also pluripotent, immortal and oncogenic [61]. ES cells like stem cells exhibit self-renew and can be programmed to differentiate [62]. ES cells are also similar to HSCs with regard to the diminished importance of the p21 response. In mouse HSCs, p21 is not essential for steady-state hematopoiesis (but could be important under conditions of IR-induced stress) [63, 64]. Relevant for this project, mouse ES proliferate rapidly and are endowed with strong replication fork maintenance properties. This is important for studying toxins that impact HSCs since replicative stress is a major contributor to their functional decline and since HSCs accumulate DNA damage as they leave a quiescent state as a direct consequence of replicative stress [65, 66]. In addition, defects in pathways that suppress broken replication forks lead to a collapse of the hematopoietic system when challenged [67]. In concurrence with these observations, we find in a nonbiased screen with ES cells that DSB repair and replication fork maintenance pathways are essential to address BQ-induced damage. Of note, mouse ES cells mutated for excision repair genes display an obvious phenotype; therefore, the absence of phenotype for these mutant cells exposed to BQ is not due to naturally diminished excision repair. Thus, BQ likely induces replicative stress that leads to DSBs to cause hematopoietic toxicity.
We propose the following model to explain benzene-induced hematopoietic toxicity. The benzene metabolite, BQ suppresses type 1 topoisomerases to inhibit replication fork restart and increase supercoiling upstream of the fork. Then PARP1-stabilized fork regression ameliorates the tension caused by supercoiling and minimizes the ATR and DNA-PKCS responses to phosphorylate RPA 32. An interesting observation is that BQ causes fewer chromosomal anomalies than either ETO or CPT at similarly toxic doses based on cell survival. It is possible that BQ is less mutagenic than ETO or CPT since it can inhibit type 1 topoisomerase nicking that would otherwise generate substrates for joining. Yet, imperfect repair or faulty maintenance of the fork would still lead to chromosomal rearrangements with the potential to develop into a hematopoietic cancer. This model proposes that people with poor genome maintenance capacity are at high risk for BQ-induced disease; of particular importance is their ability to repair DNA DSBs and maintain stabile replication forks. Our results are in concordance with reports that describe defects in HR and FA predispose people to hematopoietic cancers like MDS and AML [16, 68–70]. These individuals would likely be more susceptible to BQ toxicity further increasing their risk to develop hematopoietic disease. Furthermore, our results correspond to reports that show chemotherapeutics like ETO cause therapy-related MDS and AML (t-MDS/AML) [71, 72]. Benzene pollution would also have a greater impact on cancer patients. For such people, locating to a low-benzene environment would reduce their risk of t-MDS/AML.
MATERIALS AND METHODS
Mutant cell lines
For the experiment shown in Figure 1, we used ES cells mutated for NER (Xpa [73], Xpc [74]) MMR (Msh2 [75]), error-free postreplication repair (Rad18 [76]), FA (Fancb [77]) and nonhomologous end joining (Ku70 [78]). Complete ablation of HR is cell lethal [79]; therefore, we use null cells for several genes that contribute to, but are not essential for HR (H2ax [80], Rad52 [81], Rad54 [82]). In addition, we have cells that are partially defective for essential proteins that include a deletion of Brca2 exon 27 [26] and deletion of Brca1 exon 11 [83]. We also use cells defective for HR regulation that include mutations in the helicases Blm [84] and Recql5 [85]. We also have cells defective for the endonucleases Mus81 [86] and Ercc1 [87] and the exonuclease Trex2 [88]. All mutants were compared to their parental clone as we previously described [21].
Cell culture conditions
Mouse embryonic stem (ES) cells were cultured in Hyclone Dulbecco’s high glucose Modified Eagles Medium (GE Healthcare) with 15% fetal bovine serum (FBS) (Gemini bio-products), 2 mM glutamine (GIBCO), 30 μg/mL penicillin (Sigma), 50 μg/mL streptomycin (GIBCO), 10-4 M β-mercaptoethanol (Sigma) and 1000 units/mL leukemia inhibitory factor (Gemini bio-products). Mouse ES cells were cultured on cell culture dishes (Corning) coated with 0.1% gelatin. HeLa cells were maintained in Minimal Eagle Medium (GIBCO) with 10% FBS, 2 mM glutamine, 30 μg/mL penicillin and 50 μg/mL streptomycin. All cell lines were grown at 37°C in a 5% CO2 humidified incubator.
Dose response curves
The dose response curves were performed with a variety of mutant cells as described [21] [22]. BQ was suspended in ethanol.
Two-color fluorescent in situ hybridization (FISH)
Mouse ES cells were cultured on 10 cm plates and treated with BQ, CPT, ETO or MMC at the doses described in figure 2. The remainder of the experiments were performed as described elsewhere [77].
DNA fiber assay
For the experiment shown in Figure 3A-3C, mouse ES cells (AB2.2 and FancbΔex2) were cultured on a 6-well plate (Corning) and labeled with 25 μM 5-Iodo-2’-deoxyuridine (IdU) (Sigma) at 37°C for 20 minutes. Labeled cells were washed twice with fresh media and treated 1.5 hours with either BQ (10 μM or 60 μM) or HU (0.5 μM). Cells were again washed twice with fresh media and then labeled with 250 μM 5-Chloro-2’-deoxyuridine (CldU) at 37°C for 20 minutes. The remainder of the experiment was performed as described [25]. For the experiment shown in Figure 3D-3F, cells were labeled with IdU as described above. Labeled cells were washed twice with fresh media and then treated for 30 minutes with fresh media containing 250 μM CldU plus BQ (10 μM or 60 μM) or ETO (1 μM) or CPT (1 μM). The remainder experiments were performed as previously described [25].
Isolation of proteins on nascent DNA (iPOND)
For the experiment shown in Figure 4, mouse ES cells (AB2.2) were expanded on 15 cm plates. Cells were incubated with 10 μM 5’-ethynyl-2’-deoxyuridine (EdU) (Invitrogen) for 15 minutes. Cells incorporated with EdU were washed with fresh media and treated with agents BQ (10 μM for 1.5 hours or 60 μM for 1.5 hours), ETO (0.1 μM for 1.5 hours or 0.5 μM for 1.5 hours or 1 μM for 1.5 hours), CPT (0.1 μM for 1.5 hours or 1 μM for 1.5 hours) or HU (0.5 mM for 1.5 hours or 4 mM for 5 hours). The remainder of the experiment was performed as described [25].
Immunofluorescence (foci analysis)
For the experiment shown in Figure 5, HeLa cells were plated on LabTek chamber slides (Thermo scientific) and treated with BQ or ETO or CPT or γ-radiation (Mark1 gamma radiation source from Shepard and Associates) with or without olaparib (Sellekchem) at the doses shown in the figure. Cells were rinsed with PBS and fixed in 2% formaldehyde at room temperature for 10 minutes. Cells were then rinsed with PBS and permeabilized with 0.5% Triton X-100 at room temperature for 10 minutes. After washing with PBS for 5 minutes, three times, cells were blocked in 4% non-fat milk in PBS at room temperature for 1 hour followed by 4°C overnight incubation with a rabbit polyclonal antibody directed against 53BP1 (1:250) (A300-273A, Bethyl laboratories) and mouse monoclonal antibody directed against γH2AX (1:250). The next morning cells were washed with PBS three times and incubated with Alexa Fluor 594-conjugated goat anti-rabbit IgG and Alexa Fluor 488-conjugated goat anti-mouse IgG (both 1:1000, Molecular Probes) at RT for 1 h. After three more washes with PBS, cells were washed with PBS three times and mounted in Vectashield (Vector laboratories). Images were captured by Axioplan2 and analyzed by AxioVision software. A cell was scored as positive if it contained ≥10 foci. More than 350 cells per each sample were counted and the counting assessment was done blind.
DNA cleavage assay with topoisomerase 1
DNA cleavage assays (n=3) were performed with the Topoisomerase I assay kit from Topogen. Reaction mixtures contained 1X TGS buffer (100 mM Tris (pH 7.9), 10 mM EDTA, 1.5 M NaCl, 1% BSA, 1 mM spermidine, 50% glycerol) and 125 ng/μl supercoiled or relaxed pHOT-1 DNA. p-BQ (10-300 μM), CPT (100-500 μM) as a positive control or 100 μM ETO as a negative control and 10 U recombinant human topoisomerase I were added last and incubated at 37°C for 1 hour. Reactions were stopped by addition of 10% SDS. Next, 5 ng/μl proteinase K was added to detect clear DNA cleavages and reactions were incubated at 37°C for 30 minutes. Reactions were terminated with addition of 1X gel loading buffer (5% sarkosyl, 0.125% bromophenol blue, 25% glycerol). Samples were electrophoresed onto a 1% agarose gel (Sigma) at 55 V for 3 hours. The gel was stained with 0.5 μg/ml ethidium bromide (Fisher biotech) in 1X TAE buffer (40 mM Tris-Acetate and 1 mM EDTA (Ambion) for 30 min. followed by a 15 min. destaining in ddH2O. DNA bands were captured by Gel logic 200 imaging system (Kodak) and Carestream MI version 5.0.7.24 software (Carestream Health). This assay was performed three times with similar results.
ACKNOWLEDGMENTS
We thank all the investigators that contributed cells used for the genotoxic profile described in Figure 1 and referenced in the Materials and Methods section. We thank Laura Niedernhofer for critical review of the manuscript.
FUNDING
This work was supported by the National Institutes of Health (1 RO1 ES022054-01 and the Edward P. Evans Foundation to PH and VIR and 1 P01AG017242-17 to PH and JH).
CONFLICTS OF INTEREST
The authors have no conflict of interest.
REFFERENCES
1. Schnatter AR, Glass DC, Tang G, Irons RD and Rushton L. Myelodysplastic syndrome and benzene exposure among petroleum workers: an international pooled analysis. J Natl Cancer Inst. 2012; 104:1724-1737.
2. Hayes RB, Songnian Y, Dosemeci M and Linet M. Benzene and lymphohematopoietic malignancies in humans. Am J Ind Med. 2001; 40:117-126.
3. Rinsky RA, Smith AB, Hornung R, Filloon TG, Young RJ, Okun AH and Landrigan PJ. Benzene and leukemia. An epidemiologic risk assessment. N Engl J Med. 1987; 316:1044-1050.
4. Hartwig A. The role of DNA repair in benzene-induced carcinogenesis. Chem Biol Interact. 2010; 184:269-272.
5. Aul C, Bowen DT and Yoshida Y. Pathogenesis, etiology and epidemiology of myelodysplastic syndromes. Haematologica. 1998; 83:71-86.
6. Whitworth KW, Symanski E and Coker AL. Childhood lymphohematopoietic cancer incidence and hazardous air pollutants in southeast Texas, 1995-2004. Environ Health Perspect. 2008; 116:1576-1580.
7. Chen B, Zhao WL, Jin J, Xue YQ, Cheng X, Chen XT, Cui J, Chen ZM, Cao Q, Yang G, Yao Y, Xia HL, Tong JH, et al. Clinical and cytogenetic features of 508 Chinese patients with myelodysplastic syndrome and comparison with those in Western countries. Leukemia. 2005; 19:767-775.
8. Chatterjee T, Dixit A, Mohapatra M, Tyagi S, Gupta PK, Mishra P, Bhattacharya M, Karan AS, Pati HP, Saxena R and Choudhry VP. Clinical, haematological and histomorphological profile of adult myelodysplastic syndrome. Study of 96 cases in a single institute. Eur J Haematol. 2004; 73:93-97.
9. Lee JH, Lee JH, Shin YR, Lee JS, Kim WK, Chi HS, Park CJ, Seo EJ and Lee KH. Application of different prognostic scoring systems and comparison of the FAB and WHO classifications in Korean patients with myelodysplastic syndrome. Leukemia. 2003; 17:305-313.
10. Intragumtornchai T, Prayoonwiwat W, Swasdikul D, Suwanwela N, Chaimongkol B, Jootar S, Chansung K, Chancharunee S, Leelasiri A and Yoshida Y. Myelodysplastic syndromes in Thailand: a retrospective pathologic and clinical analysis of 117 cases. Leuk Res. 1998; 22:453-460.
11. Oguma S, Yoshida Y, Uchino H, Maekawa T, Nomura T and Mizoguchi H. Clinical characteristics of Japanese patients with primary myelodysplastic syndromes: a co-operative study based on 838 cases. Anemia Study Group of the Ministry of Health and Welfare. Leuk Res. 1995; 19:219-225.
12. Millman A, Tang D and Perera FP. Air pollution threatens the health of children in China. Pediatrics. 2008; 122:620-628.
13. Economopoulou P, Pappa V, Papageorgiou S, Dervenoulas J and Economopoulos T. Abnormalities of DNA repair mechanisms in common hematological malignancies. Leuk Lymphoma. 2011; 52:567-582.
14. Zhou T, Chen P, Gu J, Bishop AJ, Scott LM, Hasty P and Rebel VI. Potential relationship between inadequate response to DNA damage and development of myelodysplastic syndrome. International journal of molecular sciences. 2015; 16:966-989.
15. Stieglitz E and Loh ML. Genetic predispositions to childhood leukemia. Therapeutic advances in hematology. 2013; 4:270-290.
16. Mehta PA, Harris RE, Davies SM, Kim MO, Mueller R, Lampkin B, Mo J, Myers K and Smolarek TA. Numerical chromosomal changes and risk of development of myelodysplastic syndrome--acute myeloid leukemia in patients with Fanconi anemia. Cancer Genet Cytogenet. 2010; 203:180-186.
17. Lan Q, Zhang L, Shen M, Jo WJ, Vermeulen R, Li G, Vulpe C, Lim S, Ren X, Rappaport SM, Berndt SI, Yeager M, Yuenger J, et al. Large-scale evaluation of candidate genes identifies associations between DNA repair and genomic maintenance and development of benzene hematotoxicity. Carcinogenesis. 2009; 30:50-58.
18. Piwkham D, Gelfond JA, Rerkamnuaychoke B, Pakakasama S, Rebel VI, Pollock BH, Winick NJ, Collier AB, 3rd, Tomlinson GE and Beuten J. Multilocus association of genetic variants in MLL, CREBBP, EP300, and TOP2A with childhood acute lymphoblastic leukemia in Hispanics from Texas. Cancer Epidemiol Biomarkers Prev. 2011; 20:1204-1212.
19. Swinney RM, Beuten J, Collier AB, 3rd, Chen TT, Winick NJ, Pollock BH and Tomlinson GE. Polymorphisms in CYP1A1 and ethnic-specific susceptibility to acute lymphoblastic leukemia in children. Cancer Epidemiol Biomarkers Prev. 2011; 20:1537-1542.
20. Winick N, Buchanan GR and Kamen BA. Secondary acute myeloid leukemia in Hispanic children. J Clin Oncol. 1993; 11:1433.
21. Kim TM, Rebel VI and Hasty P. Defining a genotoxic profile with mouse embryonic stem cells. Exp Biol Med (Maywood). 2013; 238:285-293.
22. Marple T, Li H and Hasty P. A genotoxic screen: rapid analysis of cellular dose-response to a wide range of agents that either damage DNA or alter genome maintenance pathways. Mutat Res. 2004; 554:253-266.
23. Hu L, Kim TM, Son MY, Kim SA, Holland CL, Tateishi S, Kim DH, Yew PR, Montagna C, Dumitrache LC and Hasty P. Two replication fork maintenance pathways fuse inverted repeats to rearrange chromosomes. Nature. 2013; 501:569-572.
24. Dumitrache LC, Hu L, Son MY, Li H, Wesevich A, Scully R, Stark J and Hasty P. Trex2 enables spontaneous sister chromatid exchanges without facilitating DNA double-strand break repair. Genetics. 2011; 188:787-797.
25. Kim TM, Son MY, Dodds S, Hu L and Hasty P. Deletion of BRCA2 exon 27 causes defects in response to both stalled and collapsed replication forks. Mutat Res. 2014; 766-767:66-72.
26. Morimatsu M, Donoho G and Hasty P. Cells deleted for Brca2 COOH terminus exhibit hypersensitivity to gamma- radiation and premature senescence. Cancer Res. 1998; 58:3441-3447.
27. Marple T, Kim TM and Hasty P. Embryonic stem cells deficient for Brca2 or Blm exhibit divergent genotoxic profiles that support opposing activities during homologous recombination. Mutat Res. 2006; 602:110-120.
28. Donoho G, Brenneman MA, Cui TX, Donoviel D, Vogel H, Goodwin EH, Chen DJ and Hasty P. Deletion of Brca2 exon 27 causes hypersensitivity to DNA crosslinks, chromosomal instability, and reduced life span in mice. Genes Chromosomes Cancer. 2003; 36:317-331.
29. Lindsey RH, Jr., Bromberg KD, Felix CA and Osheroff N. 1,4-Benzoquinone is a topoisomerase II poison. Biochemistry. 2004; 43:7563-7574.
30. Sordet O, Goldman A and Pommier Y. Topoisomerase II and tubulin inhibitors both induce the formation of apoptotic topoisomerase I cleavage complexes. Mol Cancer Ther. 2006; 5:3139-3144.
31. Guenatri M, Bailly D, Maison C and Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol. 2004; 166:493-505.
32. Kim TM, Ko JH, Hu L, Kim SA, Bishop AJ, Vijg J, Montagna C and Hasty P. RAD51 mutants cause replication defects and chromosomal instability. Mol Cell Biol. 2012; 32:3663-3680.
33. Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007; 8:735-748.
34. Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, de Winter JP, Wang W and Joenje H. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004; 36:1219-1224.
35. Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ and Passmore LA. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell. 2014; 54:858-869.
36. Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J and Li L. Modularized functions of the Fanconi anemia core complex. Cell reports. 2014; 7:1849-1857.
37. Schlacher K, Wu H and Jasin M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell. 2012; 22:106-116.
38. Kim TM, Son MY, Dodds S, Hu L, Luo G and Hasty P. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 2015; 43:893-903.
39. Szekeres T, Fritzer-Szekeres M and Elford HL. The enzyme ribonucleotide reductase: target for antitumor and anti-HIV therapy. Crit Rev Clin Lab Sci. 1997; 34:503-528.
40. Carr AM, Paek AL and Weinert T. DNA replication: Failures and inverted fusions. Semin Cell Dev Biol. 2011; 22:866-874.
41. Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW and Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011; 25:1320-1327.
42. Recolin B, Van der Laan S and Maiorano D. Role of replication protein A as sensor in activation of the S-phase checkpoint in Xenopus egg extracts. Nucleic Acids Res. 2012; 40:3431-3442.
43. Anantha RW, Vassin VM and Borowiec JA. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J Biol Chem. 2007; 282:35910-35923.
44. Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A and Bonner WM. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol. 2003; 81:123-129.
45. Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM and Nussenzweig A. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010; 141:243-254.
46. Holcomb VB, Rodier F, Choi Y, Busuttil RA, Vogel H, Vijg J, Campisi J and Hasty P. Ku80 deletion suppresses spontaneous tumors and induces a p53-mediated DNA damage response. Cancer Res. 2008; 68:9497-9502.
47. Sogo JM, Lopes M and Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002; 297:599-602.
48. Berti M, Chaudhuri AR, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, Marino F, Lucic B, Biasin V, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol. 2013; 20:347-354.
49. Suvorova, II, Grigorash BB, Chuykin IA, Pospelova TV and Pospelov VA. G1 checkpoint is compromised in mouse ESCs due to functional uncoupling of p53-p21Waf1 signaling. Cell Cycle. 2016; 15:52-63.
50. Li M, He Y, Dubois W, Wu X, Shi J and Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell. 2012; 46:30-42.
51. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R and Gu W. Tumor Suppression in the Absence of p53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell. 2012; 149:1269-1283.
52. Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A and Strasser A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell reports. 2013; 3:1339-1345.
53. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS and Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356:215-221.
54. Kondratova A, Watanabe T, Marotta M, Cannon M, Segall AM, Serre D and Tanaka H. Replication fork integrity and intra-S phase checkpoint suppress gene amplification. Nucleic Acids Res. 2015; 43:2678-2690.
55. Desmarais JA, Hoffmann MJ, Bingham G, Gagou ME, Meuth M and Andrews PW. Human embryonic stem cells fail to activate CHK1 and commit to apoptosis in response to DNA replication stress. Stem Cells. 2012; 30:1385-1393.
56. Ko LJ and Prives C. p53: puzzle and paradigm. Genes Dev. 1996; 10:1054-1072.
57. Facucho-Oliveira JM, Alderson J, Spikings EC, Egginton S and St John JC. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci. 2007; 120:4025-4034.
58. Prigione A, Fauler B, Lurz R, Lehrach H and Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells. 2010; 28:721-733.
59. Varum S, Momcilovic O, Castro C, Ben-Yehudah A, Ramalho-Santos J and Navara CS. Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain. Stem Cell Res. 2009; 3:142-156.
60. Kondoh H, Lleonart ME, Bernard D and Gil J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histology and histopathology. 2007; 22:85-90.
61. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS and Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282:1145-1147.
62. Murray P and Edgar D. The topographical regulation of embryonic stem cell differentiation. Philos Trans R Soc Lond B Biol Sci. 2004; 359:1009-1020.
63. van Os R, Kamminga LM, Ausema A, Bystrykh LV, Draijer DP, van Pelt K, Dontje B and de Haan G. A Limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. 2007; 25:836-843.
64. Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A and Nimer SD. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009; 4:37-48.
65. Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, Le Beau MM, Stohr BA, Mendez J, Morrison CG and Passegue E. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014; 512:198-202.
66. Alvarez S, Diaz M, Flach J, Rodriguez-Acebes S, Lopez-Contreras AJ, Martinez D, Canamero M, Fernandez-Capetillo O, Isern J, Passegue E and Mendez J. Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat Commun. 2015; 6:8548.
67. Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, Muedder K, Klein C, Jauch A, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015; 520:549-552.
68. Aktas D, Koc A, Boduroglu K, Hicsonmez G and Tuncbilek E. Myelodysplastic syndrome associated with monosomy 7 in a child with Bloom syndrome. Cancer Genet Cytogenet. 2000; 116:44-46.
69. Ribeiro HLJ, de Oliveira RT, Maia AR, Pires Ferreira Filho LI, de Sousa JC, Heredia FF, Magalhaes SM and Pinheiro RF. Polymorphisms of DNA repair genes are related to the pathogenesis of myelodysplastic syndrome. Hematological oncology. 2014.
70. Fugger K, Mistrik M, Neelsen KJ, Yao Q, Zellweger R, Kousholt AN, Haahr P, Chu WK, Bartek J, Lopes M, Hickson ID and Sorensen CS. FBH1 Catalyzes Regression of Stalled Replication Forks. Cell reports. 2015.
71. Vardiman JW. The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: an overview with emphasis on the myeloid neoplasms. Chem Biol Interact. 2010; 184:16-20.
72. Yang D, Fu X, Zhang X, Li W and Zhang M. Therapy-related acute myeloid leukemia in patients with lymphoma: A report of four cases and review of the literature. Oncology letters. 2015; 10:3261-3265.
73. van Steeg H, Klein H, Beems RB and van Kreijl CF. Use of DNA repair-deficient XPA transgenic mice in short-term carcinogenicity testing. Toxicol Pathol. 1998; 26:742-749.
74. Cheo DL, Ruven HJ, Meira LB, Hammer RE, Burns DK, Tappe NJ, van Zeeland AA, Mullenders LH and Friedberg EC. Characterization of defective nucleotide excision repair in XPC mutant mice. Mutat Res. 1997; 374:1-9.
75. Abuin A and Bradley A. Recycling selectable markers in mouse embryonic stem cells. Mol Cell Biol. 1996; 16:1851-1856.
76. Tateishi S, Niwa H, Miyazaki J, Fujimoto S, Inoue H and Yamaizumi M. Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol Cell Biol. 2003; 23:474-481.
77. Kim TM, Ko JH, Choi YJ, Hu L and Hasty P. The phenotype of FancB-mutant mouse embryonic stem cells. Mutat Res. 2011; 712:20-27.
78. Gu Y, Jin S, Gao Y, Weaver DT and Alt FW. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc Natl Acad Sci U S A. 1997; 94:8076-8081.
79. Lim DS and Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996; 16:7133-7143.
80. Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R and Alt FW. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002; 99:8173-8178.
81. Rijkers T, Van Den Ouweland J, Morolli B, Rolink AG, Baarends WM, Van Sloun PP, Lohman PH and Pastink A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol Cell Biol. 1998; 18:6423-6429.
82. Essers J, Hendriks RW, Swagemakers SM, Troelstra C, de Wit J, Bootsma D, Hoeijmakers JH and Kanaar R. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 1997; 89:195-204.
83. Huber LJ, Yang TW, Sarkisian CJ, Master SR, Deng CX and Chodosh LA. Impaired DNA damage response in cells expressing an exon 11-deleted murine Brca1 variant that localizes to nuclear foci. Mol Cell Biol. 2001; 21:4005-4015.
84. Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA and Bradley A. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000; 26:424-429.
85. Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, Jasin M, Vogel H, Sung P and Luo G. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007; 21:3073-3084.
86. Hanada K, Budzowska M, Modesti M, Maas A, Wyman C, Essers J and Kanaar R. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. Embo J. 2006; 25:4921-4932.
87. Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH and Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. Embo J. 2001; 20:6540-6549.
88. Chen MJ, Dumitrache LC, Wangsa D, Ma SM, Padilla-Nash H, Ried T and Hasty P. Cisplatin depletes TREX2 and causes Robertsonian translocations as seen in TREX2 knockout cells. Cancer Res. 2007; 67:9077-9083.