
INTRODUCTION
Prostate cancer (PCa) is the commonest malignancy in males in the western world and accounts for approximately 10% of all male cancer-related deaths in the USA [1]. Androgen deprivation therapy (ADT) is an effective therapeutic option for advanced PCa but castration resistance invariably develops [2]. In this context, chemotherapy and other systemic therapies have limited efficacy [3]. An improved understanding of the molecular mechanisms of castration resistance can guide rational development of novel therapeutic strategies.
miRNAs play critical roles in the regulation of cell biological processes [4, 5] and are involved in the development and progression of various human malignancies including PCa [6–12]. In PCa, miRNAs have been reported to regulate cancer cell growth, survival and sensitivity to chemotherapy [6, 7, 9, 11, 13]. Although differential expression of certain miRNAs between androgen-dependent prostate cancer (ADPC) and castration-resistant prostate cancer (CRPC) clinical samples has been reported, each of those studies was performed on only a few CRPC clinical samples. The cellular functions of these differentially expressed miRNAs identified in the clinical samples were also only investigated generally for PCa cell growth instead of the development of castration resistance [14–16]. We aimed to identify novel miRNAs associated with castration resistance in PCa and to investigate the downstream pathways and molecular mechanisms. We found decreased expression of miR-4638-5p in CRPC and revealed functionally that miR-4638-5p inhibited the growth of androgen independent cancer cells and that the inhibition of miR-4638-5p promoted androgen-independent cell growth for initially androgen dependent cells. Subsequently we identified Kidins220, VEGF and the PI3K/AKT pathway as the downstream mediators of miR-4638-5p in suppressing CRPC development as well as PCa angiogenesis in vitro and in xenografted nude mice.
RESULTS
MiR-4638-5p is significantly down-regulated in CRPC compared to ADPC samples
To explore the expression change of miRNAs associated with castration resistance, we performed miRNA microarray experiments using RNA samples extracted from three CRPC and three ADPC samples (Supplementary Table S1). We found 30 underexpressed (Supplementary Table S2) and 32 overexpressed miRNAs (Supplementary Table S3) with >2 fold difference of mean value in the three CRPC samples compared with the ADPC samples (Figure 1A). 25 underexpressed and 16 overexpressed miRNAs have been previously reported in human cancers, including PCa, such as underexpression of miR-135a [17, 18], miR-143 [17, 19], miR-145 [17, 20], miR-205 [17, 21–24], miR-24-1 [17, 25], miR-146a [26, 27], miR-3607 [28], and overexpression of miR-1247 [29], miR-197 [20], miR-592 [30], miR-150 [31, 32]. Among the 30 underexpressed miRNAs in CRPC samples, miR-4638-5p, a miRNA that has not been previously reported in any human cancers, was expressed 2.4-fold higher in the ADPC than CRPC samples. Although it has not been reported in a previous publication [33], we found that miR-4638-5p was expressed 4.7-fold higher in early-stage compared to advanced disease in the miRNA microarray data from that study. In a separate miRNA microarray analysis of PCa stem cells, which are closely associated with CRPC development [34], miR-4638- 5p was the most significantly down-regulated miRNA compared with the unselected population of DU145 cells, with 12.6-fold difference (unpublished data). All these data support that reduced expression of miR-4638-5p may be responsible for a more aggressive phenotype. We further examined its expression in 30 ADPC tissues and 18 CRPC tissues by qRT-PCR. In addition to confirming the CRPC-associated downregulation of miR-4638-5p in the six samples used for microarray analysis, we found that miR-4638-5p expression level was significantly lower in CRPC compared to ADPC samples (P = 1.44 × 10-8) (Figure 1B) in this cohort of samples. We also determined the expression levels of miR-4638-5p in LNCaP, PC3, DU145 and LNCaP C4-2B PCa cell lines, consistent with our observation in the clinical samples, miR-4638- 5p expression at a much higher level in LNCaP androgen sensitive cells than the three androgen-independent PCa cell lines (Figure 1C).
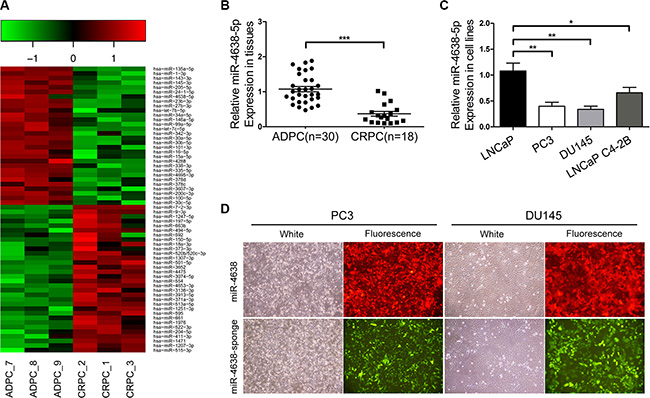
Figure 1: Downregulation of miR-4638-5p in CRPC and construction of PCa cell lines stably overexpressing/underexpressing miR-4638-5p. (A) Heatmap of top miRNAs with differential expression in ADPC and CRPC tissues. Pseudo-color was used to represent intensity scale of miRNAs expression from the array analysis. Green and red denote low and high expression, respectively. (B) The expression levels of miR-4638-5p in 18 CRPC and 30 ADPC samples were detected by qRT-PCR. (C) The expression levels of miR-4638-5p in LNCaP, PC3, DU145 and LNCaP C4-2B cells were detected by qRT-PCR. * indicates P < 0.05, ** indicates P < 0.01 and *** indicates P < 0.001 by Student’s t-test. (D) The expression of RFP and EGFP from the pCDH-miR-4638-5p (miR-4638) and the pCDH-miR-4638-5p sponge (miR-4638 sponge, to inhibit miR-4638-5p) were observed under the fluorescence microscope after that PCa cells were infected with lentivirus 72 hours under the same multiplicity of infection. (original magnification, ×100).
MiR-4638-5p suppresses androgen-independent PCa cell growth in vitro and in vivo
We then explored the potential biological functions of miR-4638-5p in cancer cell growth in relation to androgen independence. We constructed androgen independent PC3 and DU145 PCa cell lines stably overexpressing or underexpressing miR-4638-5p (with pCDH-miR-4638-5p sponge) (Figure 1D). As expected, miR-4638-5p expression were increased by approximately 25- and 18-fold respectively in PC3 and DU145 cells transfected with pCDH-miR-4638-5p or reduced by 5- and 3-fold, respectively, in PC3 and DU145 cells transfected with pCDH-miR-4638-5p sponge, compared with each cell line transfected with empty-vector control (all P < 0.05) (Supplementary Figure S1). In addition, miR-4638- 5p evidently inhibited the reporter activity of pGL3-miR-4638-5p sensor reporter, further indicating that the miR-4638-5p lentiviral expression constructs in PCa cells were functional (Supplementary Figure S2). Overexpressing miR-4638-5p significantly reduced cell growth rate in comparison with cells transduced with the empty pCDH-vector control (P = 5.56 × 10-3 and P = 4.42 × 10-2for PC3 and DU145 cells, respectively) (Figure 2A), while the growth rate of PC3 and DU145 cells stably expressing miR-4638-5p sponge was significantly enhanced compared to cells transduced with the pCDH-vector control (P = 4.34 × 10-2 and P = 3.37 × 10-2 for PC3 and DU145 cells, respectively) (Figure 2A) as detected by CCK-8 assay. We also performed colony formation assay using those transfected cells and found that miR-4638-5p expressing PC3 and DU145 cells had lower colony formation abilities than cells transduced with the pCDH-vector control (P = 4.75 × 10-2 and P = 3.27 × 10-2 for PC3 and DU145 cells, respectively) and these cells stably expressing miR-4638-5p sponge showed opposite findings (P = 2.18 × 10-2 and P = 4.62 × 10-2 for PC3 and DU145 cells, respectively) (Figures 2B and 2C). To further explore whether the decline in cell growth rate of the above miR-4638-5p overexpressing CRPC cells is caused by the induction of apoptosis in addition to the reduced proliferative capacity, we analyzed the proportion of apoptotic cell by flow cytometry. The results demonstrated that the percentage of total apoptotic cells were significantly increased after miR-4638 mimic transfection in comparison to the scramble RNA negative control (P = 2.93 × 10-2 and P = 1.14 × 10-2 for PC3 and DU145 cells, respectively). In addition, PC3 and DU145 cells transfected with miR-4638 inhibitor significantly reduced cell apoptosis compared with the cells transfected with the scramble RNA negative control (P = 2.68 × 10-3 and P = 1.27 × 10-2, respectively) and miR-4638 mimic (Figure 2D and 2E). Subsequently, we investigated the effect on cell proliferation by reducing miR-4638- 5p expression level in androgen sensitive LNCaP cells. LNCaP cells cultured in non/very low androgen medium with charcoal-stripped serum have very limited proliferation capacity. However, when miR-4638-5p was inhibited by stably expressing miR-4638-5p sponge, LNCaP cells continuously proliferate and at day 5 there were significantly (P = 1.24 × 10-2) more cells than LNCaP transfected with the pCDH-vector control in androgen-stripped cell culture medium (Figure 2F), indicating that miR-4638-5p inhibits certain cellular pathways which are required for PCa cell proliferation when androgen is not available.
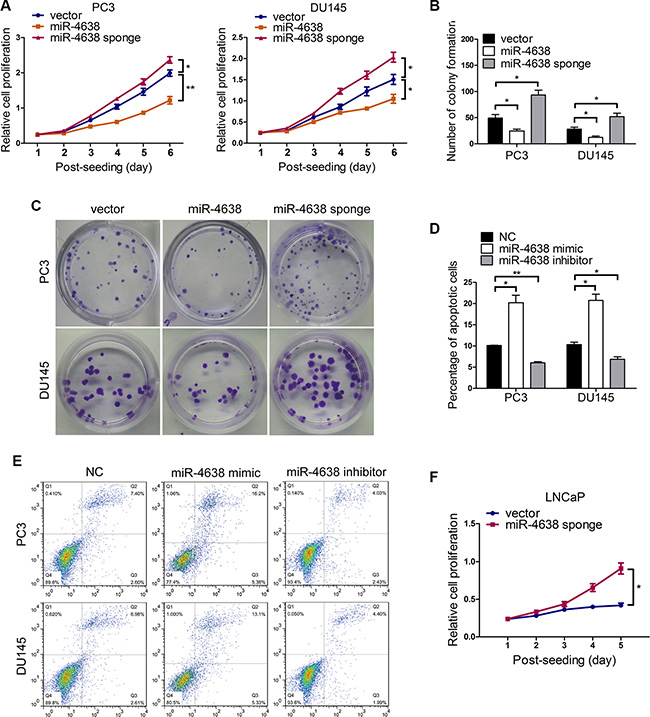
Figure 2: The effects of miR-4638-5p on cell proliferation, colony formation, apoptosis and the development of castration resistance. (A) Cell proliferation results of PC3 and DU145 cells transduced with the pCDH-miR-4638-5p (miR-4638) or the pCDH-miR-4638-5p sponge (miR-4638 sponge, to inhibit miR-4638-5p), determined by a Cell Counting Kit-8 assay (CCK-8). Data represent mean ± SD determined from three independent experiments (n = 3), each with four technical replicates. Representative images from CCK-8 assay, showing the reduced or increased proliferative capacity of both PC3 and DU145 cells after stably overexpressing miR- 4638-5p or miR-4638-5p sponge, respectively, in comparison with cells transduced with the pCDH-vector controls (vector). (B) Colony formation ability of PC3 and DU145 cells transduced with the pCDH-miR-4638-5p (miR-4638) or the pCDH-miR-4638-5p (miR-4638 sponge). Bars represent the mean ± SD from three independent experiments (n = 3), each with three technical replicates. (C) Representative images showing the increased or decreased colony formation ability of both PC3 and DU145 cells after stably overexpressing miR-4638-5p or miR-4638-5p sponge, respectively, in comparison with cells transduced with the pCDH-vector controls (vector) after 2 weeks of seeding (original magnification, ×100). (D) Cell apoptosis assay was performed by flow cytometry analysis. The forced expression of miR-4638-5p (miR-4638 mimic) significantly increased the sensitivity of PC3 and DU145 cells to apoptosis, compared with the scramble RNA negative control (NC). The percentage of total apoptotic cells decreased in response to miR-4638-5p inhibitor (miR-4638 inhibitor) transfection. Bars represent the mean ± SD from three independent experiments (n = 3), each with three technical replicates. (E) Representative FACS analysis images. (F) LNCaP cells stably expressing miR-4638-5p sponge proliferate significantly faster than the pCDH-vector control cells in androgen stripped cell culture medium. Bars represent the mean ± SD from three independent experiments (n = 3), each with four technical replicates. * indicates P < 0.05 and ** indicates P < 0.01 by Student’s t-test.
We then investigated the impact of miR-4638- 5p expression change on androgen independent tumor growth in vivo. We injected the PC3 cells overexpressing miR- 4638-5p into nude mice and observed a delay in tumor onset and a marked decrease in tumor growth when compared with the pCDH-vector control transfected cells that both the tumor volume and weight were significantly different at day 28 (P = 1.48 × 10-3 and P = 1.60 × 10-3, respectively). The opposite result was observed in cells transfected with pCDH-miR-4638-5p sponge compared to the pCDH-vector control (P = 4.36 × 10-2 and 3.23 × 10-2 for tumor volume and weight respectively) (Figures 3A–3C). In addition, Ki-67 positive cells were significantly reduced in PC3 cells transduced with pCDH-miR-4638-5p (P = 3.96 × 10- 2) and opposite result was observed in the PC3 cells transduced with pCDH-miR-4638-5p sponge (P = 3.51 × 10-2) (Figure 3D and 3E), suggesting that miR-4638-5p reduced tumor growth in the nude mouse model through inhibiting cell proliferation.
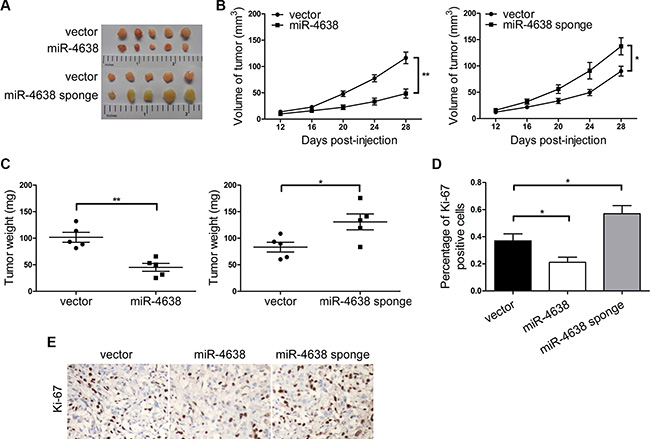
Figure 3: miR-4638-5p inhibits tumor growth in nude mice. (A) Tumors grew in nude mice removed at day 28 after xenograft. (B) Overexpression of miR-4638-5p inhibited tumor growth and inhibition of miR-4638-5p using miR-4638-5p sponge promoted tumor growth of xenograpted PC3 cells based on two-dimensional caliper measurements of the tumor size. Data represent mean ± SD, each group with five tumours (n = 5). Two independent experiments were performed and similar results were obtained. (C) Overexpression of miR- 4638- 5p inhibited tumor growth and inhibition of miR-4638-5p using miR-4638-5p sponge promoted tumor growth of xenograpted PC3 cells based on the weight of those removed tumours. (D) miR-4638-5p overexpression and inhibition significantly decreased (P = 3.96 × 10-2) and increased (P = 3.51 × 10-2), respectively, Ki-67 expression detected by immunostaining in tumours from xonografted PC3 cells. (E) Representative immunostaining images (original magnification, ×100). * indicates P < 0.05 and ** indicates P < 0.01 by Student’s t-test, respectively.
MiR-4638-5p directly targets Kidins220
Since miRNAs usually inhibit the translation of proteins via non-perfect pairing of six to eight nucleotides in length [35] or induce the degradation of target mRNAs in the case of perfect complementarity with the potential binding sites of target genes [36], bioinformatics analysis with several programs including TargetScan, RNAhybrid, Findtar, and Pita, was performed to predict the putative miR-4638-5p targets. Based on non-perfect complementarity with the seed sequences of miR-4638-5p, some putative binding sites were predicted in the 3′UTRs of six potential target genes, USP49, CSAD, SEC61A2, TATDN3, C10orf12 and Kidins220. Subsequent luciferase reporter assay using 3′-UTR region of the six candidate genes confirmed that the promoter activity of four out of the six genes were significantly inhibited by miR-4638-5p (P < 0.05 for all) (Figure 4A) in HEK293T cells. However, only Kidins220 protein expression was significantly inhibited by miR-4638-5p in PC3 and DU145 cells as determined by Western blotting analysis (Figure 4B), while the expression changes of the other three proteins were not significant. As expected, knockdown of miR-4638-5p increased the expression of Kidins220 protein in those cells (Figure 4B). Moreover, luciferase reporter assay showed that miR-4638-5p inhibited the activity of Kidins220 3′-UTR WT in a dose-dependent fashion in HEK293T cells (Figure 4C). As expected, overexpression of miR-4638-5p using 50 nM and 100 nM miR-4638 mimics also suppressed Kidins220 protein expression in PC3 and DU145 cells in a dose-dependent manner (Figure 4D), further supporting that Kidins220 is the gene regulated by miR-4638-5p. To confirm the specificity of miR-4638-5p in targeting Kidins220, we performed 3′-UTR mutagenesis analysis, where we mutated the putative miR-4638-5p binding site in the Kidins220 3′-UTR WT (Figure 4E) and found that the mutant mimic lacking the seed sequences reduced the inhibitory effect of miR-4638-5p on luciferase expression (P < 0.05; Figure 4F). In addition, the mutant miR-4638 mimic designed to match the Kidins220 3′-UTR mut (Figure 4E) exhibited a strong inhibitory effect on the mutant reporter (P = 1.28 × 10- 2; Figure 4F). These results indicated that miR-4638-5p directly targets Kidins220 with specific binding sites at the seed sequences of 3′- UTR.
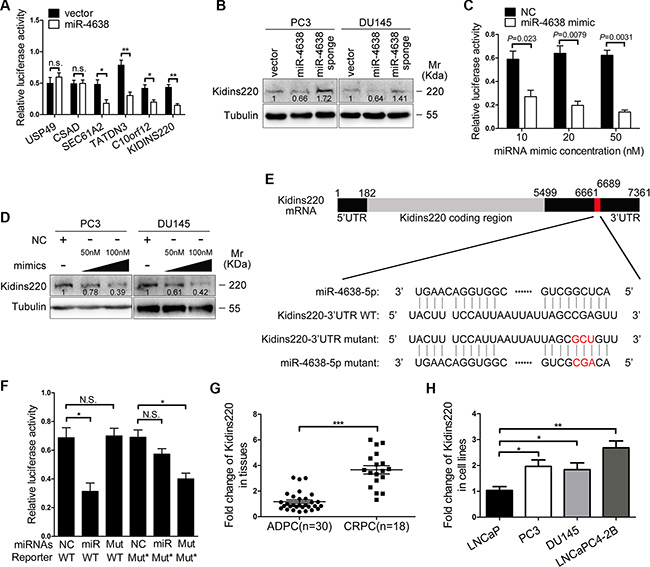
Figure 4: The regulation of Kidins220 by miR-4638-5p and the association of Kidins220 expression with CRPC. (A) Luciferase activity under the control of 3′UTR of candidate genes in HEK293T cells transfected with the pCDH-miR-4638-5p (miR-4638) and the pCDH-vector control (vector). (B) The expression of Kidins220 protein in PC3 and DU145 cells transduced with the pCDH-miR-4638-5p (miR-4638) or the pCDH-miR-4638-5p sponge (miR-4638 sponge). (C) Luciferase activity under the control of Kidins220 3′UTR in HEK293T cells transfected with increasing amounts (10, 20 and 50 nM) of miR-4638 mimics for 48 hours. NC: scramble RNA negative control. (D) The expression of Kidins220 protein in PC3 and DU145 cells transfected with increasing amounts (50 and 100 nM) of miR-4638 mimics for 72 hour. (E) Schematic illustration of the putative seed sequence of miR-4638-5p within Kidins220 3′UTR and mutagenesis of binding site in the 3′UTR of Kidins220 or miR-4638 mimic. (F) Effect of seed mutagenesis or mutation of the putative binding site on the Kidins220 3′UTR activity. Kidins220 wild type (WT) and mutant 3′UTR construct (Mut*) were co-transfected with the scramble RNA negative control (NC), natural (miR) or mutant miR-4638 mimic (Mut) into HEK293T cells. After co-transfection for 48 h, HEK293T cells were assayed for luciferase activity. In (A) (C) (F), the data represent the mean ± SD from three independent experiments (n = 3), each experiment containing four technical replicates. (G) Kidins220 expression in 18 CRPC and 30 ADPC samples quantitated by qRT-PCR. (H) Kidins220 expression in LNCaP, PC3, Du145 and LNCaP C4-2B cells. Relative quantities of mRNA expression are represented on the y-axis. *, ** and *** indicate P < 0.05, P < 0.01 and P < 0.001 respectively. n.s.: not significant.
MiR-4638-5p may suppress CRPC cell growth by inhibiting Kidins220 expression
To explore whether miR-4638-5p may suppress CRPC development by inhibiting Kidins220 expression, we firstly examined the expression of Kidins220 in the 30 ADPC and 18 CRPC samples as well as PCa cell lines using qRT-PCR, we found that Kidins220 expression was significantly much higher in CRPC tissues compared with ADPC tissues (P = 3.10 × 10-7; Figure 4G), which was reversely correlated with miR-4638-5p in those two groups of clinical samples. In addition, the mRNA of Kidins220 was also detected at higher levels in the androgen independent LNCaP C4-2B, PC3 and DU145 cells than in the androgen sensitive LNCaP cells (P < 0.05; Figure 4H). To further determine the cellular effect of Kidins220 in CRPC cells, we knocked down Kidins220 in PC3 and DU145 cells. We tested three Kidins220 shRNAs, which all showed knockdown effect at the protein levels, and selected shRNA (S-2), which achieved the best knockdown effect (Figure 5A) to establish stable Kidins220 knockdown cells (Figure 5B). As expected, compared with the pCDH-vector control cells, Kidins220-knockdown PC3 and DU145 cells showed a reduction in cell proliferation and the increase of apoptotic cells in vitro (P < 0.05; Figures 5C–5E) and reduced tumor growth in xenographed nude mice (P < 0.05; Figure 5F–5H), mimicking the effects of miR-4638-5p overexpression.
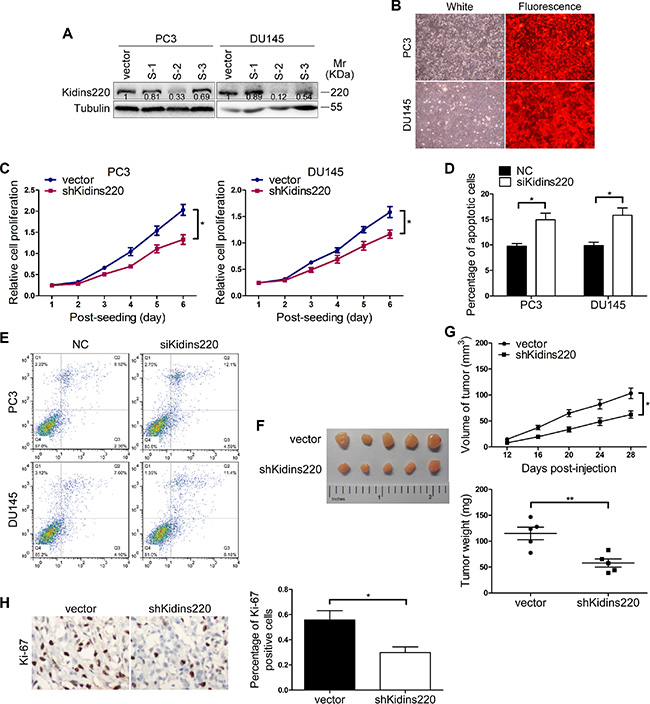
Figure 5: Kidins220 knockdown mimics the effort of miR-4638-5p overexpression on tumor cell growth. (A) Western blot analysis of Kidins220 expression levels of PC3 and DU145 cells transduced with shRNA1-3 (S1-3). (B) The expression of RFP were observed under the fluorescence microscope 72 hours after PC3 and DU145 cells were infected with pCDH-shKidins220 (shKidins220) or the pCDH-vector controls (vector) under the same multiplicity of infection. (original magnification, ×100). (C) Cell proliferation results of PC3 and DU145 cells transduced with the pCDH-shKidins220 (shKidins220) or the pCDH-vector controls (vector), determined by a Cell Counting Kit-8 (CCK-8) assay. Data represent mean ± SD determined from three independent experiments (n = 3), each with four technical replicates. Representative images from CCK-8 assay, showing the reduced proliferative capacity of both PC3 and DU145 cells after stably knockdown of Kidins220, in comparison with cells transduced with the pCDH-vector controls. (D) Cell apoptosis assay was performed by flow cytometry analysis. The knockdown of Kidins220 (siKidins220) significantly increased the sensitivity of PC3 and DU145 cells to apoptosis, compared with the scramble RNA negative control (NC). Bars represent the mean ± SD from three independent experiments (n = 3), each with three technical replicates. (E) Representative flow cytometry images. (F) Tumors grew in nude mice removed at day 28 after xenograft. (G) Knockdown of Kidins220 inhibited tumor growth of xenograpted PC3 cells based on two-dimensional caliper measurements of the tumor size (upper panel) and the weight of those removed tumours (lower panel). Data represent mean ± SD, each group with five tumours (n = 5). Two independent experiments were performed and similar results were obtained. (H) Kidins220 knockdown significantly decreased (P = 2.08×10-2) Ki-67 expression detected by immunostaining in tumours from xonografted PC3 cells with representative immunostaining images (original magnification, ×100) on the left. * indicates P < 0.05, and ** indicates P < 0.01 by Student’s t-test, respectively.
Kidins220, mediating the effect of miR-4638-5p downregulation, promotes PCa neo-angiogenesis
As Kidins220 have been reported to activate VEGFR [37], which play a key role in cancer neo-angiogenesis, we also investigated the role of Kidins220 in PCa angiogenesis. Microtubule formation assay was performed with HUVECs and conditioned medium collected from culture supernatant fluid of PC3 cells 48 hours after Kidins220 knockdown by shRNA. We found that knockdown of Kidins220 in PC3 cells decreased microtubule formation ability of HUVECs in vitro (P = 1.14 × 10-2; Figure 6A). Vasculogenic mimicry analysis was also performed with PC3 cells with and without Kidins220 knockdown and we found that knockdown of Kidins220 reduced 62% mimicry formation of PC3 cells (P = 2.83 × 10-3; Figure 6B). We also determined whether knockdown of Kidins220 in the androgen-independent PC3 cells would inhibit angiogenesis in vivo by measuring the final hemoglobin content using Drabkin’s reagent kit in the xenografts and found that hemoglobin content in the tumor xenografts of cells with Kidins220 knockdown was significantly (P = 1.96 × 10-2) decreased compared with control cells transfected with non-targetting shRNA (Figure 6C). As expected, we detected significantly decreased VEGF expression in xenografts of Kidins220 knockdown PC3 cells by immunostaining (P = 6.06 × 10-3; Figure 6D).
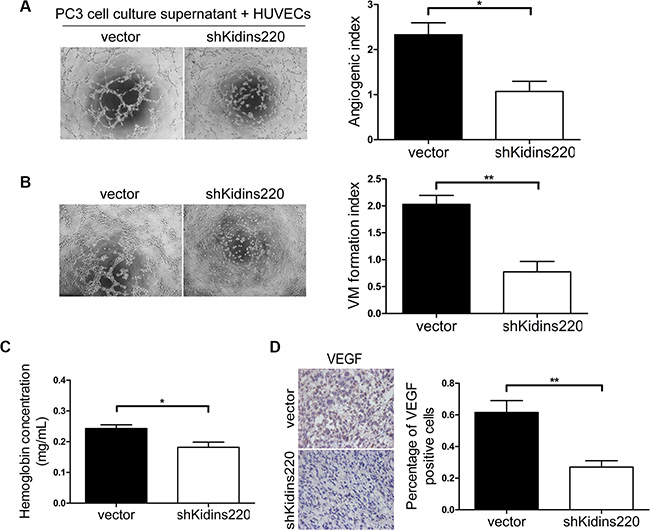
Figure 6: Kidins220 knockdown inhibits the microtubule and vasculogenic mimicry formation of PCa cells and angiogenesis in vivo. (A) Matrigel assay of microtubule formation. Tube formation assay was performed with HUVECs incubated in culture supernatant fluid of PC3 cells 48 hours after Kidins220 knockdown or not. The photographs of microtubules were captured at 6 hour post seeding (original magnification, ×100). The quantification data represent the mean ± SD from three independent experiments (n = 3), each experiment containing four technical replicates. (B) Vasculogenic mimicry formation assay was performed with PC3 cells. The mimicry formation ability of PC3 cells transduced with the pCDH-shKidins220 (shKidins220) was significantly decreased, compared to the cells transduced with the pCDH-vector controls (vector). Representative images of mimicry were captured at 6 hour post seeding (original magnification, ×100) on the left. The data represent the mean ± SD from three independent experiments (n = 3), each experiment containing four technical replicates. (C) The hemoglobin level of the Matrigel plugs in the PC3 xonografted tumours with Kidins220 knockdown was significantly decreased. Data represent mean ± SD, each group with five tumours (n = 5). Three independent experiments were performed with similar results. (D) Kidins220 knockdown significantly decreased VEGF expression as detected by immunostaining in tumours from xonografted PC3 cells (P = 6.06 × 10-3) with representative immunostaining images (original magnification, ×100) on the left. * indicate P < 0.05 and ** indicate P < 0.01 by Student’s t-test, respectively.
Following the finding that Kidins220 influences PCa angiogenesis, we explored whether miR-4638- 5p suppresses PCa angiogenesis both by inhibiting microtubule formation of endothelial cells through VEGF paracrine secretion and the formation of vasculogenic mimicry by PCa cells. The microtubule formation ability of HUVECs incubated with culture supernatant of PC3 cells overexpressing miR-4638-5p was significantly (P = 9.92 × 10-3) weaker than cells incubated with culture supernatant of those PCa cells transduced with the pCDH-vector control. HUVECs incubated with culture supernatant of PC3 cells transduced with pCDH-miR-4638-5p sponge showed significantly (P = 4.74 × 10-2) higher angiogenesis ability compared with the pCDH-vector control (Figures 7A), indicating that miR-4638-5p may block the microtubule formation ability of HUVECs through VEGF paracrine secretion in PC3 cells. We also performed vasculogenic mimicry analysis of PC3 cells with different level of miR-4638-5p. Vasculogenic mimicry formation ability of PC3 cells transduced with the pCDH-miR-4638-5p or pCDH-miR-4638-5p sponge were significantly decreased (P = 9.42 × 10-4) or increased (P = 4.79 × 10-2), compared to the cells transduced with the pCDH-vector control (Figures 7B). Furthermore, our in vivo experiments using tumor xenografts from PC3 cells showed that the final hemoglobin content in tumor xenografts of PC3 cells transduced with the pCDH-miR-4638-5p was significantly (P = 4.39 × 10-3) decreased compared with the empty vector transduced control cells (Figure 7C). The opposite effect was observed with cells transduced with pCDH-miR-4638-5p sponge in comparison to the control cells (P = 7.87 × 10-3) (Figure 7C). MiR-4638-5p overexpression and inhibition also significantly decreased (P = 1.60 × 10-3) and increased (P = 4.51 × 10-2), respectively, VEGF expression in xonografted PC3 cells as detected by immunostaining (Figure 7D). Therefore, the phenotype observed upon miR-4638-5p overexpression and Kidins220 knockdown were similar, suggesting that miR-4638-5p may target Kidins220 to regulate not only androgen independent tumor growth but also PCa angiogenesis both through stimulating endothelial cells and forming vasculogenic mimicry.
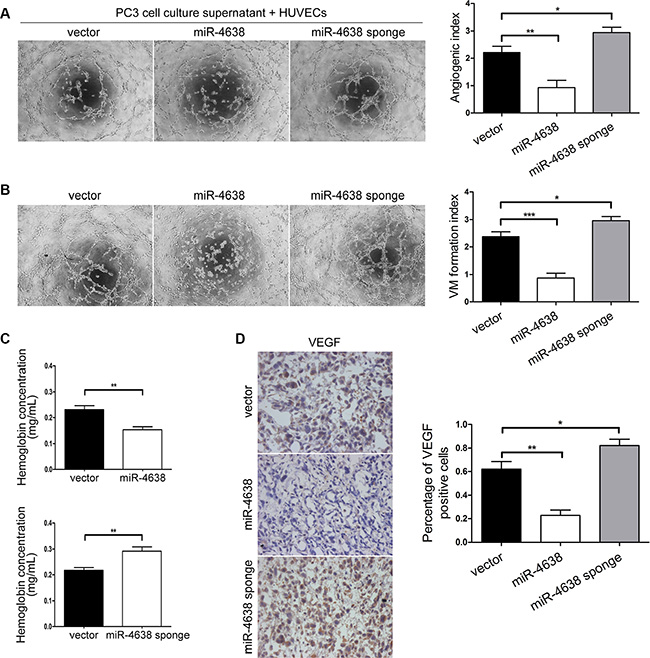
Figure 7: miR-4638-5p inhibits angiogenesis in vitro and in vivo and mimicry formation of PCa cells. (A) Matrigel assay of microtubule formation. Tube formation assay was performed with HUVECs cells incubated in culture supernatant fluid of PC3 cells 48 hours after transfection with the pCDH-miR-4638-5p (miR-4638) or the pCDH-miR-4638-5p sponge (miR-4638 sponge). The photographs of microtubules were captured at 6 hour post seeding (original magnification, ×100). The quantification data represent the mean ± SD from three independent experiments (n = 3), each experiment containing four technical replicates. (B) Representative mimicry formation images (original magnification, ×100) and bar chart showing that vasculogenic mimicry formation ability of PC3 cells transduced with the pCDH-miR-4638- 5p (miR-4638) or the pCDH-miR-4638-5p sponge (miR-4638 sponge) were significantly decreased or increased, respectively compared to the cells transduced with the pCDH-vector controls (vector). The data represent the mean ± SD from three independent experiments (n = 3), each experiment containing four technical replicates. (C) The hemoglobin level of the Matrigel plugs in the PC3 xonografted tumours with miR-4638-5p overexpression and inhibition was significantly decreased (P = 4.39 × 10-3) and increased (P = 7.87 × 10-3), respectively. Data represent mean ± SD, each group with five tumours (n = 5). Three independent experiments were performed with similar results. (D) miR-4638-5p overexpression and inhibition also significantly decreased (P = 1.60 × 10-3) and increased (P = 4.51 × 10- 2), respectively, VEGF expression detected by immunostaining in tumours from xonografted PC3 cells. Representative immunostaining images (original magnification, ×100) on the left. *, ** and *** indicate P < 0.05, P < 0.01 and P < 0.001 by Student’s t-test, respectively.
PI3K/AKT pathway may mediate KIDINS220-induced androgen independent PCa growth and angiogenesis
To further investigate cellular signaling pathway regulated by miR-4638-5p and shKidins220, we performed Western blotting for candidate downstream genes responsible for CRPC and angiogenesis, in the PC3 and DU145 cells transfected with the pCDH-miR-4638-5p, pCDH-miR-4638-5p sponge or empty pCDH-vector control. We found that stable transduction with the pCDH-miR-4638-5p or pCDH-miR-4638-5p sponge resulted in the reduction or increase, respectively, of VEGF as well as phosphorylated forms of PI3K and AKT (p-PI3K and p-AKT, respectively) (Figure 8A). Consistent with the results of miR-4638-5p manipulating, Kidins220 knockdown by lentivirus mediated shKidins220 transfection similarly inhibited the VEGF, p-PI3K and p-AKT protein expression (Figure 8B). To further investigate whether PI3K/AKT pathway mediates Kidins220-induced tumor growth and angiogenesis, we knocked down AKT with shRNA (Figure 8C and 8D). As expected, compared with the pCDH-vector transfected control cells, AKT-knockdown PC3 and DU145 cells showed reduction in cancer cell growth (P < 0.05 for all; Figure 8E and Figure 9A–9B) and angiogenesis (P < 0.01 for all; Figure 8F and Figure 9C–9D) in vitro and in vivo, similar to the phenotype observed upon Kidins220-knockdown. Meanwhile, vasculogenic mimicry formation ability of PC3 cells transduced with shAKT was significantly decreased (P = 4.22 × 10-3), compared to the cells transduced with the pCDH-vector control (Figure 8G). Taken together, these results suggest that PI3K/AKT pathway may mediate Kidins220-induced angiogenesis and vasculogenic mimicry in addition to PCa cell proliferation and castration resistance.
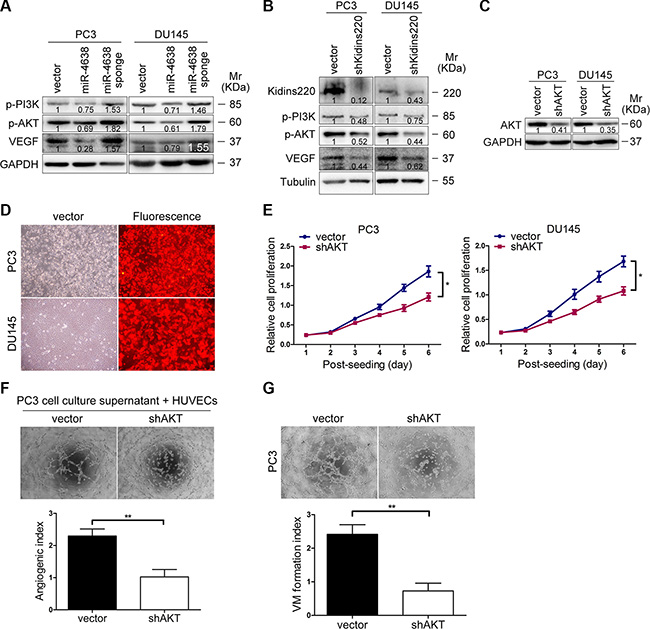
Figure 8: PI3K/AKT signaling pathway mediates Kidins220-induced tumor growth, angiogenesis and mimicry formation of PCa cells. (A) Western blot analysis of VEGF and phosphorylation levels of PI3K, AKT (p-PI3K and p-AKT, respectively) in PC3 and DU145 cells transduced with pCDH-miR-4638-5p (miR-4638) or pCDH-miR-4638-5p sponge (miR-4638 sponge). Numbers labeled under the bands were the relative intensities of the bands after calibrating for loading with the house-keeping protein. The relative value of proteins in the vector group was considered as ‘1’. (B) Western blot analysis of total Kidins220, VEGF and phosphorylation levels of PI3K and AKT (p-PI3K and p-AKT, respectively) in PC3 and DU145 cells transduced with the pCDH-shKidins220 and the controls. Numbers labeled under the bands were the relative intensities of the bands after calibrating for loading with the house-keeping protein. The relative value of proteins in the vector group was considered as ‘1’. (C) Western blot analysis of total AKT levels in PC3 and DU145 cells transduced with the pCDH-shAKT or not. Numbers labeled under the bands were the relative intensities of the bands after calibrating for loading with the house-keeping protein. The relative value of proteins in the vector group was considered as ‘1’. (D) The expression of RFP observed under the fluorescence microscope 72 hours after PC3 and DU145 cells were infected with the pCDH-shAKT and the pCDH-vector control under the same multiplicity of infection. (original magnification, ×100). (E) Cell proliferation results of PC3 and DU145 cells transduced with the pCDH-shAKT (shAKT) or the pCDH-vector controls (vector), showing the reduced proliferative capacity of both PC3 and DU145 cells after stably underexpressing AKT. Data represent mean ± SD determined from three independent experiments (n = 3), each with four technical replicates. (F) Matrigel analysis of microtubule formation. Tube formation assay was performed with HUVECs cells incubated in culture supernatant fluid of PC3 cells 48 hours after AKT knockdown or not. The photographs of microtubules were captured at 6 hour post seeding (original magnification, ×100) on the top. Quantification of results was performed subsequently. The data represent the mean ± SD from three independent experiments (n = 3), each experiment containing four technical replicates. (G) Vasculogenic mimicry formation assay was performed with PC3 cells. The mimicry formation ability of PC3 cells transduced with the pCDH-shAKT (shAKT) was significantly decreased, compared to the cells transduced with the pCDH-vector controls (vector) with representative images of mimicry captured at 6 hour post seeding (original magnification, ×100) on the top. The data represent the mean ± SD from three independent experiments (n = 3), each experiment containing four technical replicates. * indicate P < 0.05 and ** indicate P < 0.01 by Student’s t-test, respectively.
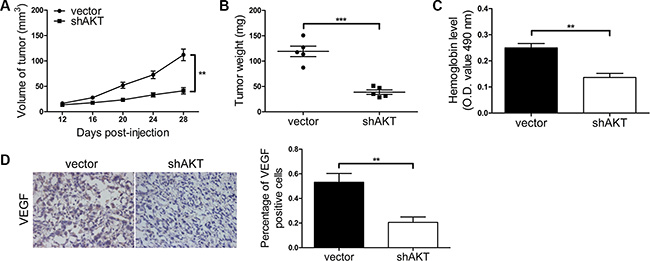
Figure 9: PI3K/AKT signaling pathway mediates Kidins220-induced tumorigenesis and angiogenesis in vivo. (A–B) AKT knockdown reduced tumorigenesis in vivo. PC3 cells were transduced with the pCDH-shAKT (shAKT) or the pCDH-vector control (vector) for 72 hour and further re-suspended in serum-free medium. The sizes of tumors from nude mice were determined by two-dimensional caliper measurements. Scatter plots represent the weights of independent tumors from different groups. Data represent mean ± SD, each group with five tumors (n = 5). Two independent experiments were performed and similar results were obtained. (C) The hemoglobin level of the Matrigel plugs was determined with O.D. value at 540 nm. Data represent mean ± SD and each group with five tumors (n = 5). Three independent experiments were performed and similar results were obtained. (D) AKT knockdown significantly decreased VEGF expression detected by immunostaining in tumors from xonografted PC3 cells (P = 6.29 × 10-3) with representative immunostaining images (original magnification, ×100) shown on the left. ** and *** indicate P < 0.01 and P < 0.001 by Student’s t-test, respectively.
DISCUSSION
CRPC is the main cause of mortality from PCa. Our current knowledge on CRPC development is mainly focused on androgen metabolism and androgen receptor pathways [38]. Identification of other molecular changes associated with CRPC will greatly help us to control this common and lethal form of male malignancy. Alterations of many miRNAs have been reported in PCa development, progression as well as response to therapies [39]. The role of miRNAs in androgen signaling and CRPC development has also been investigated in cell lines and animal models [40]. Differential expression of miRNAs between CRPC and ADPC samples have also been reported [41–44]. Here, using miRNA microarray analysis, we identified a number of differentially expressed miRNAs between Chinese ADPCs and CRPCs. While many of them have been previously studied, miR-4638-5p has not been investigated previously in any human cancers. The investigation of the down-regulation of miR-4638- 5p in PCa was also supported by its down-regulation in advanced disease [33] and PCa stem cells from separate miRNA microarray data. MiR-4638-5p was the mostly underexpressed miRNA in isolated stem cells compared to non-stem DU145 cells. PCa stem cell are closely associated with castration resistance, that PCa stem cell markers have been associated with the development of CRPC, such as CD166 [45], Nanog [46, 47], Bmi-1 [48] and Sox2 [49, 50]. We also further confirmed the downregulation of miR-4638-5p in CRPC samples using a larger series of clinical samples and demonstrated both through in vitro and in vivo studies that miR-4638-5p play a significant role in suppressing androgen independent PCa cell growth.
The function of miR-4638-5p is currently unknown; the only published report on miR-4638-5p was from a bioinformatics analysis of potential intronic and 3′UTR targeted miRNAs from known cardiac marker genes, where miR-4638-5p has been supposed to regulate cardiac marker gene MYH-7, which encodes beta-myosin heavy chain and is expressed in cardiac and skeletal muscle tissues [51]. Here we demonstrated that miR-4638-5p is a putative tumor suppressor, which suppresses PCa growth, angiogenesis and castration resistance development through regulating Kidins220, VEGFR and PI3K/AKT pathway. As miRNA has been developed for therapeutic use [52], miR-4638-5p may represent a novel target for CRPC treatment. The miR-4638-5p level change, as a driver, may occurs before the clinical features of CRPC, thus it has the potential to be developed into a biomarker for the onset of CRPC before PSA increase or other clinical progressions.
In the search for genes/proteins regulated by miR-4638-5p, we identified Kidins220 as a gene whose expression was downregulated post-transcriptionally by miR-4638-5p through targeting the Kidins220 3′- UTR seed sequences. In consistent to miR-4638-5p under-expression, we demonstrated that CRPC samples overexpressed Kidins220 compared to ADPC samples and knockdown of Kidins220 generated similar effects as miR- 4638-5p overexpression in CRPC cells.
Kidins220 (kinase D–interacting substrate of 220 kDa), also known as ankyrin repeat-rich membrane spanning protein (ARMS), is a recently identified tetra-spanning-transmembrane protein abundantly expressed in the developing and adult neural tissues [53, 54]. Previous studies of Kidins220 were mainly focused on its roles in neural development and Kidins220, as a downstream substrate of protein kinase, neurotrophin and ephrin, regulates neuronal differentiation and survival [55–58]. Kidins220 also expresses outside the nervous system. It interacts with the microtubule and actin cytoskeleton, modulating cell plasticity and migration and is important for cardiovascular development and certain immune function [58–60]. Although most of the above cellular features are associated with cancer development and progression, research of Kidins220 in human malignancies is very limited. Kidins220 overexpression has been reported in melanoma and it has been shown to contribute to tumor formation by activating MEK/ERK signaling pathway and consequently preventing transformed melanocytes from the stress-induced apoptosis [61]. Expression of Kidins220 has also been reported in neuroblastoma tumours, where it stabilizes nerve growth factor-induced survival signalling through MAPK/ERK signalling [62]. Both of these studies demonstrated that Kidins220 protects cells for survival through the MAPK/ERK signalling pathway. However, the role of Kidins220 in PCa has not been reported. In this study we demonstrated that Kidins220 promoted CRPC cell growth and tumor angiogenesis and enhanced VEGFR, PI3K and AKT activity in CRPC cancer cells. These findings suggest that, in addition to the above reported roles, Kidins220 impacts on PI3K/AKT and VEGF/VEGFR pathways to promote PCa angiogenesis and the development of castration resistance. MiR- 4638- 5p, to our knowledge, is also the first identified miRNA to regulate Kidins220. Overall, these novel findings increase our understanding of the cellular and biological functions of miR-4638-5p and Kidins220 as well as reveal new mechanisms underlying the development of CRPC.
The PI3K/AKT signaling is a critical pathway in cell proliferation, survival, neovascularization and tumor growth [63, 64]. Previous studies have identified that PI3K/AKT signaling plays a key role in the development and maintenance of CRPC [65, 66]. Deregulation of PI3K/AKT signaling occurs in almost all cases of advanced CRPC [67]. Recent studies have revealed a direct link between PI3K/AKT and AR signaling pathways that they interplay during the development of castration resistance [68, 69]. In addition, the activation of PI3K/AKT signaling pathway promotes castration resistance and the inhibition of AR function accelerates the development of CRPC in PTEN deficient mice [70]. As mentioned above, the known function of Kidins220 in cancer is to protect cells for survival when they are under stress [61, 62]. In PCa, Kidins220 may activate PI3K/AKT signaling to sustain cancer cell growth under the stress of androgen deprivation and lead to CRPC. Inhibiting a single target in the PI3K/AKT signaling pathway have shown limited clinical efficacy and simultaneously inhibition of multiple targets in series or parallel have demonstrated greater promise in the treatment of CRPC [65]. As Kidins220 regulates both PI3K and AKT activities, targeting miRNA-4638- 5p and/or Kidins220 offers another promising therapeutic approach for the management of CRPC.
In this study, we provided evidence that Kidins220 modulated angiogenesis of PCa cells via regulation of both the VEGF/VEGFR2 and PI3K/AKT signaling pathway. It has been reported that Kidins220 interacts with VEGFR2 and VEGFR3 in mouse endothelial cells and modulates VEGF signaling through promoting the interaction between VEGF and VEGFR for cardiovascular development [37]. VEGF is the main angiogenic growth factor [71] and PI3K/AKT signaling interplay with VEGF level to regulate angiogenesis [64], which is critical for tumor development when cancer reaches to a certain size. Numerous studies have demonstrated that Hif-1α activation could increase the expression and secretion of VEGF via PI3K/AKT/mTOR signaling pathway [72–74]. Meanwhile, VEGF itself can effectively activate PI3K/AKT signaling pathway, stimulating blood vessel in autocrine and paracrine manner. Therefore, the observation that Kidins220 regulated angiogenesis in human cancers via activation of VEGF and PI3K/AKT signaling is not surprising, but this has not been investigated before. In this study, we also showed for the first time that Kidins220 through VEGF and PI3K/AKT signaling regulates both neo-vascularization from endothelial cells and the formation of vasculogenic mimicry of CRPC cells for tumor angiogenesis. Vasculogenic mimicry (VM) phenomenon provides a new pathway for tumor blood supply independent of endothelial cells [75]. The tumor cells possessing mimic vessel display highly malignant characteristics of invasive growth, dedifferentiation and metastasis [76]. The ability of vasculogenic mimicry formation of cancer cells may be an additional mechanism for them to access sufficient nutrition and growth factors for cell survival and proliferation under adverse stressful environment, such as androgen deprivation in the case of CRPC. Based on our results, a model of miR-4638-5p and Kidins220 functions for cell survival and angiogenesis has been proposed (Figure 10).
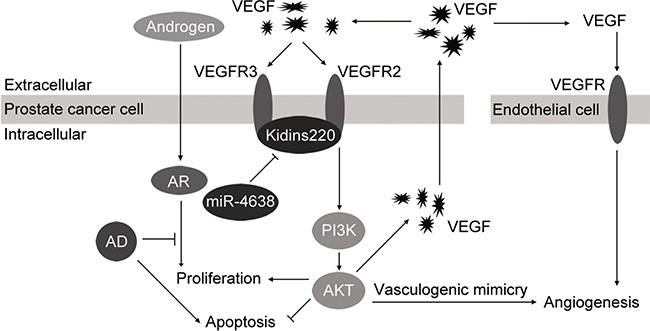
Figure 10: Proposed model of miR-4638-5p in control cell growth and angiogenesis in prostate cancer cell. Kidins220, regulated by miR-4638-5p, binds to the VEGFR2 and VEGFR3 and promotes interaction between VEGF and VEGFR as a co-receptor (auxiliary receptors) of VEGFR, so that VEGF/PI3K/AKT signaling pathway is activated to promote proliferation and mimicry formation and suppress apoptosis of PCa cells. AKT activation also consequently increases the expression level and secretion of VEGF, which stimulates angiogenesis of endothelial cells. Therefore, miR-4638-5p inhibits these cellular process by downregulating its target gene Kidins220.
While angiogenesis is critical for cancer progression and anti-angiogenesis therapy has been extensively tested with certain success in treating CRPC [77], the role of angiogenesis in CRPC development has not been reported. Therefore, the role of accelerated angiogenesis by miRNA-4638-5p down-regulation and the consequently deregulation of the above molecular pathways in castration development is unclear. Angiogenesis may provide oxygen, nutrition and other growth factors to help the survival of cancer cells under stress of androgen depletion during CRPC development. Neuroendocrine PCa is AR negative and with poor prognosis, therefore, the development of neuroendocrine PCa has been considered as a mechanism of CRPC [78]. Kidins220 is critical for neuronal differentiation and survival [37, 55, 57–59]. As we have demonstrated in vitro and in vivo that Kidins220 promote tumor angiogenesis through activation of PI3K/AKT and VEGF signalling, Kiddins220 may also involved in both neuroendocrine differentiation and angiogenesis of PCa cells. Both neuroendocrine differentiation and vasculogenic mimicry formation are cellular changes of losing epithelial features of PCa cells. Kiddins220 may coordinate neuroendocrine differentiation and angiogenesis, together with regulating PI3K/AKT signaling, promoting the development of castration resistance. Further investigations are required.
In summary, we identified a novel tumor suppressor miRNA, miR-4638-5p, which is downregulated in CRPC, and dissected its target genes and downstream cell signaling pathways. We revealed for the first time that miR-4638-5p regulates Kidins220 mRNA by targeted degradation and consequently suppression of VEGF/VEGFR and PI3K/AKT in CRPC cells, and impact on tumor growth and angiogenesis using in vitro and in vivo models. While the role of PI3K/AKT pathway in CRPC initiation is well established, the role of VEGF signaling and angiogenesis in CRPC development warrants further investigations. These findings provide new molecular insights into the development of CRPC and can guide the development of novel approaches targeting the miR- 4638- 5p/Kidins220/VEGF/VEGFR/PI3K/AKT axis to prevent or treat CRPC.
MATERIAL AND METHODS
Human prostate tissue specimens
Fresh frozen tissue from 30 ADPC and 18 CRPC patients were obtained from Wuxi No. 2 People’s Hospital affiliated with Nanjing Medical University. The 18 CRPC patients were selected based on their increased serum prostate-specific antigen (PSA) level during androgen-deprivation therapy and the cancer tissues were obtained through transurethral prostatic resection to relieve the symptom of urinary obstruction. The 30 ADPC samples were derived from radical prostatectomy in treatment-naive patients. Clinical stage of disease was determined by histopathology, magnetic resonance image and radio-nucleotide bone scans according to the 2002 TNM classification of prostate carcinoma. The clinic-pathological features of all study patients are summarized in Supplementary Table S1. All these samples contained more than 60% tumor by pathological examination. The use of these human PCa tissues for this study was approved by the institutional ethics review board of Nanjing Medical University.
Cell lines
Human umbilical vein endothelial cells (HUVECs) were maintained in EBM-2 culture media (LONZA, Allendale, NJ, USA) as previously described [79]. DU145 human androgen independent PCa cell line and HEK293T were maintained in DMEM, and androgen dependent LNCaP, androgen independent LNCaP C4-2B subline and PC3 human PCa cell lines was cultured in RPMI 1640, all supplemented with 10% fetal bovine serum (FBS), 100 units/ml of penicillin and 100 μg/ml of streptomycin.
MiRNA microarray analysis
Total RNA was isolated using TRizol (Invitrogen) and purified with RNeasy mini kit (QIAGEN) according to manufacturer’s instructions. RNA quality and quantity was measured using NanoDrop spectrophotometer (ND-1000, NanoDrop Technologies) and RNA Integrity was determined by gel electrophoresis. 2.5 μg total RNA samples were labeled using the miRCURYTM Array Power Labeling kit (Exiqon, Vedbaek, Denmark) following the manufacturer’s instruction. Microarray images were acquired using a Genepix 4000B scanner (Axon Instruments, Union City, CA, USA), processed and analyzed with Genepix Pro 6.0 software (Axon Instruments).
Plasmids and miRNA mimics
The miR-4638-5p (Invitrogen, Shanghai, China), Kidings220 shRNA (Invitrogen) and AKT shRNA, (Invitrogen), were cloned into the lentiviral vector pCDH-CMV-MCS-EF1-tRFP with modification as previously described [80] and a miR-4638-5p sponge (Invitrogen) as described below was cloned into another lentiviral vector pCDH-CMV-MCS-EF1-copGFP (System Bioscience, CA, USA) for stable miR-4638-5p, Kidings220 and AKT overexpression and/or knockdown studies together with the empty lentiviral vector (pCDH-vector control). Synthetic miR-4638-5p mimic (miR-4638 mimic, RIBOBIO, Guangzhou, China), miR-4638-5p inhibitor (miR-4638 inhibitor, RIBOBIO), Kidings220 siRNA (siKidins220, RIBOBIO) and corresponding scramble RNA negative controls (RIBOBIO) were applied for transient overexpression or knockdown analyses. pGL3 luciferase reporter control plasmid (pGL3-Control, Promega, Shanghai, China), pGL3-Luc-Kidins220-3′-UTR WT and pGL3-Luc-Kidins220-3′-UTR Mut reporter plasmids as described below, miR-4638 mimic and miR-4638 mutation mimic (RIBOBIO) were used for luciferase reporter assay to investigate Kidins220 promoter activity.
To generate miR-4638-5p lentiviral constructs (pCDH-miR-4638-5p), the sequence of miR-4638-5p (5′-ACTCGGCTGCGGTGGACAAGT-3′), obtained from the miRBase, and its complementary sequence with two mutation sites (5′-TCTTGTCCACCGGAGCCGAGT-3′) were inserted between the stem-loop structure and the flanking sequence of pre-miR-30 as previously described [80], to obtain the precursor stem-loops of miR-4638-5p, which was synthesized by Invitrogen and used as the PCR template sequence to be amplified by the following PCR primers designed using Primer Premier 5.0 software, forward: 5′-CAGAAGGCTCG AGAAGGTATATTGCTGTTGACAGTGAGCG-3′ (with Xho I sequence underlined) and Reverse: 5′-CTAAAG TGACCCCTTGAATTCCGAGGCAGTAGGCA-3′ (with EcoR I sequence underlined). The amplified precursor stem-loops of miR-4638-5p was cloned into the Xho I and EcoR I sites of the lentiviral vector pCDH-CMV-MCS-EF1-tRFP.
For the purpose of long-term stable downregulation of miR-4638-5p, miRNA sponge was produced by linking six complementary sequences of miR-4638-5p, germinated four consecutive restrictive mutations with five linker sequences to generate the miR-4638-5p sponge: 5′-ACTTGTCCAGGCGAGCCGAGTCGATACTTGTCC AGGCGAGCCGAGTACCGGTACTTGTCCAGGCGAG CCGAGTCGATACTTGTCCAGGCGAGCCGAGTCGA TACTTGTCCAGGCGAGCCGAGTACCGGTACTTGT CCAGGCGAGCCGAGT-3′, which was synthesized by Invitrogen. The miR-4638-5p sponge was cloned from the template plasmid pUC57-AMP+ into the EcoR I and BamH I sites of the lentiviral vector pCDH-CMV-MCS-EF1-copGFP to generate miR-4638-5p sponge lentiviral constructs (pCDH-miR-4638-5p sponge).
To further conform that the miR-4638-5p expression construct is functional in prostate cancer cells, the pGL3-miR-4638-5p sensor reporter (pGL3-miR-4638 Sensor for short) was produced by linking three tandem miR-4638-5p seed binding sequences and inserted into the downstream of luciferase sequence in the pGL3-Control plasmid. The sequence is as following: (forward) 5′-ACTTGTCC ACCGCAGCCGAGTAAGCTTACTTGTCCACCGCAG CCGAGTAAGCTTACTTGTCCACCGCAGCCGAGT-3′ and (reverse) 5′-ACTCGGCTGCGGTGGACAAGTAA GCTTACTCGGCTGCGGTGGACAAGTAAGCTTACT CGGCTGCGGTGGACAAGT-3′.
To stably knockdown Kidins220, the following three shRNAs for Kidins220 were designed using bioinformatics software including siDirect, Integrated DNA Technologies, Learning with Clontech and SDS (siRNA Design Software).
shRNA-01: 5′-ATATGCAAGGGAGTATCTCCT-3′;
shRNA-02: 5′-AGTTAACCCCACATTTCAGTA-3′;
shRNA-03: 5′-ATTCTCATTCTGAATAGTGGT-3′. The sequence of shRNAs of Kidins220 and their complementary sequences with one mutation site (5′-TGGA GATACTCCCTTGCATAT-3′, 5′-AACTGAAATGTGG GGTTAACT-3′, 5′-TCCACTATTCAGAATGAGAAT-3′) were inserted between the stem-loop structure and the flanking sequence of pre-miR-30 as previously described [80]. The shRNA sequences were amplified by PCR and cloned into the Xho I and EcoR I sites of the lentiviral vector pCDH-CMV-MCS-EF1-tRFP to produce pCDH-shKidins220. The same approach was used to make the AKT shRNA lentiviral construct, pCDH-shAKT, to knockdown AKT.
Cell transfections
For transient transfection of miR-4638 mimic, miR-4638 inhibitor, promoter constructs and siRNA of Kidins220, cells of 50%-60% confluence were transfected using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instruction.
To generate stable expression cell lines, the virus-packaging cells HEK293T (1 × 106) were seeded in a 10 cm dish one day before co-transfection with pCDH-miR-4638-5p, pCDH-miR-4638-5p sponge, pCDH-shKidins220 and pCDH-shAKT, packaging plasmid psPAX2 and envelope plasmid pMD2.G. 48 hours after transfection, the virus containing supernatant was collected and filtered using 0.45 μm filter to generate Lentivirus-pCDH-miR-4638-5p, Lentivirus-pCDH-miR-4638-5p sponge, Lentivirus-pCDH-shKidins220 and Lentivirus-pCDH-shAKT. The viral titer was determined by observing the expression of red or enhanced green fluorescent protein (RFP or EGFP). DU145 and PC3 cells were infected with these lentivirus under the same multiplicity of infection. The expression of RFP and EGFP was observed under the fluorescence microscope after 72 hours.
Quantitative real-time PCR analysis of miRNA and mRNAs
Total RNA was extracted from PCa tissues and cultured cells using the TRizol method (Invitrogen) and subjected to the TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA) or the Promega Reverse Transcription Kit (Promega, Madison, WI) to obtain cDNA. TaqMan MicroRNA (Assay ID: 462195_mat for has-miR-4638- 5p) and gene expression (Assay ID: Hs01057000_m1 for Kidins220) assays (Applied Biosystems) were performed in accordance with the manufacturer’s instructions using a 7500 Real-time PCR System (Applied Biosystems). The expression of U6 snRNA (Assay ID: 001973) was used as a control for miRNA expression and GAPDH (Assay ID: Hs02758991_g1) was used as a control for gene expression. The relative expressions were calculated using the 2-ΔΔCt method.
Cell proliferation assay
Cell Count Kit-8 (CCK-8) (Dojindo Molecular Technologies, Tokyo, Japan) was used to evaluate cell proliferation. PC3 and DU145 cells stably transfected with pCDH-miR-4638-5p, pCDH-miR-4638-5p sponge, pCDH-shKidins220, pCDH-shAKT and pCDH-vector controls were seeded into 96-well plates at density of 2 × 103 cells per well and cultured for 1 day, 2 days, 3 days, 4 days, 5 days and 6 days. Each well of cells were then cultured in 20 μl of CCK-8 solution in addition to the 200 μl of culture medium for 1.5 hours at 37°C. Absorbance at 450 nm was measured using a Vmax microplate spectrophotometer (Molecular Devices). LNCaP cells transfected with pCDH-miR-4638-5p sponge and pCDH-vector control were seeded into 96-well plates (also 2 × 103 cells per well) in the RPMI 1640 medium supplemented with 10% charcoal-stripped FBS and cultured for 1 day, 2 days, 3 days, 4 days and 5 days. Then the cell proliferation ability was evaluated using CCK-8 solution as described above.
Colony formation assay
Cells were seeded into 12-well plates at a low density of 100 cells per plate and cultured for 14 days. The cells were then fixed with 95% methanol and pigmented with 0.1% crystal violet. The number of colonies with >10 cells observed under low magnification microscope were counted.
Flow cytometry analysis of cell apoptosis
For the cell apoptosis analysis, PC3 and DU145 cells, 48 hour after transfection with miR-4638 mimic, miR-4638 inhibitor, siKidins220 and the scramble RNA negative controls (RIBOBIO), were collected and resuspended to 1 × 105 cells in 195 μL of buffer. Then 5 μl Annexin V-FITC and 10 μl PI staining (Beyotime, Shanghai, China) were added into the cell mix and incubated for 15 minutes in the dark at room temperature. The apoptosis rates were determined using the flow cytometry-BD FACS Calibur (BD Biosciences) and Flowjo software one hour after staining.
Matrigel tube formation assay
Microtubule formation assay was performed as previously described [81]. Briefly, 96-well plates were coated with 60 μl Matrigel solution (BD Bioscience, New Bedford, MA, USA) diluted at 1:1 in cell culture medium at 4°C. The plate was allowed to solidify for one hour at 37°C before cell seeding. The conditioned medium was collected from supernatant fluid of 48-hour cultured PCa cells with different transfections and filtered using 0.45 μm filter. HUVECs in 150 μl conditioned medium diluted at 2:1 in EBM-2 culture media without FBS, were seeded at density of 1 × 104 cells per well. After incubation at 37°C for 6 hours, four randomly selected fields of each well were photographed. The angiogenesis index was calculated using the formula as previously described [81].
Vasculogenic mimicry analysis
96-well plates were coated with 60 μl Matrigel solution diluted as a 1:1 mixture of High Concentration Matrigel (BD Bioscience) and culture medium without FBS, penicillin and streptomycin solution at 4°C. The plate was allowed to polymerize for one hour at 37°C. PC3 cells, stably transfected with pCDH-miR-4638-5p, pCDH-miR-4638-5p sponge, pCDH-shKidins220, pCDH-shAKT and pCDH-vector controls in RPMI 1640 medium without FBS, were seeded at density of 1.5 × 104 per well. After incubation at 37°C for 6 hours, three randomly selected fields of each well were photographed. The vasculogenic mimicry index was calculated using the same method as microtubule formation assay described above.
Tumorigenicity analysis and Matrigel plug assay for angiogenesis analysis in nude mice
BALB/C nu/nu male athymic mice (3-4 weeks old) were purchased from Model Animal Research Center of Nanjing University (Nanjing, China). All animal experiments were evaluated and approved by the institutional ethics review board of National Institute of Health Guide for the Care and Use of Laboratory Animals and Wuxi No. 2 People’s Hospital. The treated cells (PC3 cells infected with lentivirus) were harvested at a sub-confluent density, washed with phosphate-buffered saline (PBS) and re-suspended in serum-free medium. 2 × 107cells in 500 μl of serum-free medium were mixed with 500 μl of High Concentration Matrigel (BD Biosciences) and 100 μl mixture (2 × 106 cells) was immediately injected subcutaneously into the left or right flanks of nude mice. The tumor size was measured every 4 days with a two-dimensional caliper and tumor volume was calculated using the formula: 0.52 × length × width2. At day 28 after injection, mice were sacrificed and the xenograft tumors were surgically removed. Tumor samples were collected to make fresh frozen as well as formalin-fixed and paraffin-embedded sections for gene expression analysis. The hemoglobin content of the Matrigel plug was determined using Drabkin’s reagent kit according to the manufacturer’s instruction (Sigma-Aldrich) for tumor angiogenesis analysis. The final hemoglobin level was determined by spectrophotometric analysis at 540 nm.
Immunohistochemistry staining
Formalin-fixed paraffin-embedded tissue samples from nude mice were immunostained using ABC method with antibodies against VEGF (Cat No. sc-152, 1:50 dilution) (Santa Cruz, CA, USA) and Ki-67 (Cat No. ab15580, 1:100 dilution) (Abcam, Cambridge, UK) according to the manufacturer’s instructions as previously described [82].
Luciferase reporter assay
Human Kidins220 3′-UTR sequence was obtained from NCBI gene database [83] and synthesized by Invitrogen. The full length Kidins220 3′-UTR sequence was amplified and cloned into pGL3-Control downstream of the luciferase gene to generate the plasmid pGL3-Luc-Kidins220-3′-UTR WT (Kidins220-3′-UTR WT). Meanwhile, pGL3-Luc-Kidins220-3′-UTR Mut (Kidins220-3′-UTR mut) was generated from Kidins220-3′-UTR WT by changing three base pairs of the miR- 4638-5p seed-sequence. The wild-type or mutant reporter plasmids were co-transfected with the miR-4638- 5p or negative control and renilla vector pRL- TK into HEK293T cells (about 1 × 105) at 50%–60% confluence using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. After 48 hours, relative luciferase activity was measured using the Promega Dual-Luciferase Reporter (DLR) Assay System. At least three independent transfections were done for each experiment condition.
Western blot analysis
Proteins were separated by 8% and 10% SDS polyacrylamide gels and electrophoretically transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked overnight with 5% non-fat dried milk and incubated overnight with antibodies. Anti-Kidins220 rabbit polyclonal antibody (PAb) (Cat No. ab97345) was purchased from Abcam Inc (Abcam, Cambridge, UK). Anti-PI3K rabbit monoclonal antibody (MAb) (Cat No. ♯4257), anti-phospho-PI3K rabbit PAb (Cat No. ♯4228), anti-AKT rabbit PAb (Cat No. ♯9272) and anti-phospho-AKT (Cat No. ♯4060) were all purchased from Cell Signaling Technologies (Beverly, MA, USA). Anti-VEGF rabbit PAb (Cat No. sc-152), anti-GAPDH mouse MAb (Cat No. sc-47724), anti-a-Tubulin mouse MAb (Cat No. sc-23948) and horseradish peroxidase (HRP) conjugated goat anti-rabbit (Cat No. sc-2004) or anti-mouse (Cat No. sc-2005) secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The signals were detected by enhanced chemiluminescence (Millipore).
Statistical analysis
All experiments were performed at least in triplicates unless otherwise mentioned. Numerical data were expressed as mean ± SD. Two group comparisons were analyzed by two-sided Student’s t-test. P < 0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Chun Lu at the Department of Pathogenic Microorganisms, Nanjing Medical University, China for friendly providing pGL3-Control luciferase reporter plasmid, renilla vector pRL-TK, lentiviral vector pCDH-CMV-MCS-EF1-tRFP/pCDH-CMV-MCS-EF1-copGFP, packaging vector psPAX2, envelope vector pMD2.G. and lentiviral vector pCDH-shAKT. We also thank Jacek Marzec for technical support.
CONFLICTS OF INTREST
All authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by the National Natural Science Foundation of China (Grant No. 81272831), Natural Science Foundation of Jiangsu Province (Grant No. BK2010577), Jiangsu Province’s Outstanding Medical Academic Leader program (Grant No. RC201178) and Orchid.
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9–29.
2. Egan A, Dong Y, Zhang H, Qi Y, Balk SP, Sartor O. Castration-resistant prostate cancer: adaptive responses in the androgen axis. Cancer Treat Rev. 2014; 40:426–33.
3. Katzenwadel A, Wolf P. Androgen deprivation of prostate cancer: Leading to a therapeutic dead end. Cancer letters. 2015; 367:12–7.
4. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004; 303:83–6.
5. Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005; 6:376–85.
6. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature reviews Cancer. 2006; 6:857–66.
7. Kojima K, Fujita Y, Nozawa Y, Deguchi T, Ito M. MiR- 34a attenuates paclitaxel-resistance of hormone-refractory prostate cancer PC3 cells through direct and indirect mechanisms. The Prostate. 2010; 70:1501–12.
8. Galardi S, Mercatelli N, Farace MG, Ciafre SA. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res. 2011; 39:3892–902.
9. Li T, Li D, Sha J, Sun P, Huang Y. MicroRNA-21 directly targets MARCKS and promotes apoptosis resistance and invasion in prostate cancer cells. Biochemical and biophysical research communications. 2009; 383:280–5.
10. Xu B, Niu X, Zhang X, Tao J, Wu D, Wang Z, Li P, Zhang W, Wu H, Feng N, Hua L, Wang X. miR-143 decreases prostate cancer cells proliferation and migration and enhances their sensitivity to docetaxel through suppression of KRAS. Mol Cell Biochem. 2011; 350:207–13.
11. Huang S, Guo W, Tang Y, Ren D, Zou X, Peng X. miR- 143 and miR-145 inhibit stem cell characteristics of PC-3 prostate cancer cells. Oncology reports. 2012; 28:1831–7.
12. Aakula A, Kohonen P, Leivonen SK, Makela R, Hintsanen P, Mpindi JP, Martens-Uzunova E, Aittokallio T, Jenster G, Perala M, Kallioniemi O, Ostling P. Systematic Identification of MicroRNAs That Impact on Proliferation of Prostate Cancer Cells and Display Changed Expression in Tumor Tissue. European urology. 2015.
13. Xu B, Niu X, Zhang X, Tao J, Wu D, Wang Z, Li P, Zhang W, Wu H, Feng N, Wang Z, Hua L, Wang X. miR-143 decreases prostate cancer cells proliferation and migration and enhances their sensitivity to docetaxel through suppression of KRAS. Molecular and cellular biochemistry. 2011; 350:207–13.
14. Liu D, Tao T, Xu B, Chen S, Liu C, Zhang L, Lu K, Huang Y, Jiang L, Zhang X, Huang X, Han C, Chen M. MiR-361–5p acts as a tumor suppressor in prostate cancer by targeting signal transducer and activator of transcription-6(STAT6). Biochem Biophys Res Commun. 2014; 445:151–6.
15. Lu K, Liu C, Tao T, Zhang X, Zhang L, Sun C, Wang Y, Chen S, Xu B, Chen M. MicroRNA-19a regulates proliferation and apoptosis of castration-resistant prostate cancer cells by targeting BTG1. FEBS Lett. 2015; 589:1485–90.
16. Xu B, Wang N, Wang X, Tong N, Shao N, Tao J, Li P, Niu X, Feng N, Zhang L, Hua L, Wang Z, Chen M. MiR- 146a suppresses tumor growth and progression by targeting EGFR pathway and in a p-ERK-dependent manner in castration-resistant prostate cancer. Prostate. 2012; 72:1171–8.
17. Coarfa C, Fiskus W, Eedunuri VK, Rajapakshe K, Foley C, Chew SA, Shah SS, Geng C, Shou J, Mohamed JS, O’Malley BW, Mitsiades N. Comprehensive proteomic profiling identifies the androgen receptor axis and other signaling pathways as targets of microRNAs suppressed in metastatic prostate cancer. Oncogene. 2015.
18. Kroiss A, Vincent S, Decaussin-Petrucci M, Meugnier E, Viallet J, Ruffion A, Chalmel F, Samarut J, Allioli N. Androgen-regulated microRNA-135a decreases prostate cancer cell migration and invasion through downregulating ROCK1 and ROCK2. Oncogene. 2015; 34:2846–55.
19. Zhou P, Chen WG, Li XW. MicroRNA-143 acts as a tumor suppressor by targeting hexokinase 2 in human prostate cancer. American journal of cancer research. 2015; 5:2056–63.
20. Zhu J, Wang S, Zhang W, Qiu J, Shan Y, Yang D, Shen B. Screening key microRNAs for castration-resistant prostate cancer based on miRNA/mRNA functional synergistic network. Oncotarget. 2015; 6:43819–30. doi: 10.18632/oncotarget.6102.
21. De Cola A, Volpe S, Budani MC, Ferracin M, Lattanzio R, Turdo A, D’Agostino D, Capone E, Stassi G, Todaro M, Di Ilio C, Sala G, Piantelli M, et al. miR-205–5p-mediated downregulation of ErbB/HER receptors in breast cancer stem cells results in targeted therapy resistance. Cell death & disease. 2015; 6:e1823.
22. He HC, Han ZD, Dai QS, Ling XH, Fu X, Lin ZY, Deng YH, Qin GQ, Cai C, Chen JH, Jiang FN, Liu X, Zhong WD. Global analysis of the differentially expressed miRNAs of prostate cancer in Chinese patients. BMC genomics. 2013; 14:757.
23. Pennati M, Lopergolo A, Profumo V, De Cesare M, Sbarra S, Valdagni R, Zaffaroni N, Gandellini P, Folini M. miR-205 impairs the autophagic flux and enhances cisplatin cytotoxicity in castration-resistant prostate cancer cells. Biochemical pharmacology. 2014; 87:579–97.
24. Tsuchiyama K, Ito H, Taga M, Naganuma S, Oshinoya Y, Nagano K, Yokoyama O, Itoh H. Expression of microRNAs associated with Gleason grading system in prostate cancer: miR-182–5p is a useful marker for high grade prostate cancer. The Prostate. 2013; 73:827–34.
25. Goto Y, Kojima S, Nishikawa R, Enokida H, Chiyomaru T, Kinoshita T, Nakagawa M, Naya Y, Ichikawa T, Seki N. The microRNA-23b/27b/24–1 cluster is a disease progression marker and tumor suppressor in prostate cancer. Oncotarget. 2014; 5:7748–59. doi: 10.18632/oncotarget.2294.
26. Sun Q, Zhao X, Liu X, Wang Y, Huang J, Jiang B, Chen Q, Yu J. miR-146a functions as a tumor suppressor in prostate cancer by targeting Rac1. The Prostate. 2014; 74:1613–21.
27. Xu B, Huang Y, Niu X, Tao T, Jiang L, Tong N, Chen S, Liu N, Zhu W, Chen M. Hsa-miR-146a-5p modulates androgen-independent prostate cancer cells apoptosis by targeting ROCK1. The Prostate. 2015; 75:1896–903.
28. Saini S, Majid S, Shahryari V, Tabatabai ZL, Arora S, Yamamura S, Tanaka Y, Dahiya R, Deng G. Regulation of SRC kinases by microRNA-3607 located in a frequently deleted locus in prostate cancer. Molecular cancer therapeutics. 2014; 13:1952–63.
29. Scaravilli M, Porkka KP, Brofeldt A, Annala M, Tammela TL, Jenster GW, Nykter M, Visakorpi T. MiR- 1247–5p is overexpressed in castration resistant prostate cancer and targets MYCBP2. The Prostate. 2015; 75:798–805.
30. Lv Z, Rao P, Li W. MiR-592 represses FOXO3 expression and promotes the proliferation of prostate cancer cells. International journal of clinical and experimental medicine. 2015; 8:15246–53.
31. Dezhong L, Xiaoyi Z, Xianlian L, Hongyan Z, Guohua Z, Bo S, Shenglei Z, Lian Z. miR-150 is a factor of survival in prostate cancer patients. Journal of BUON. 2015; 20:173–9.
32. Liu DZ, Zhang HY, Long XL, Zou SL, Zhang XY, Han GY, Cui ZG. MIR-150 promotes prostate cancer stem cell development via suppressing p27Kip1. European review for medical and pharmacological sciences. 2015; 19:4344–52.
33. Wang N, Li Q, Feng NH, Cheng G, Guan ZL, Wang Y, Qin C, Yin CJ, Hua LX. miR-205 is frequently downregulated in prostate cancer and acts as a tumor suppressor by inhibiting tumor growth. Asian J Androl. 2013; 15:735–41.
34. Li P, Yang R, Gao WQ. Contributions of epithelial-mesenchymal transition and cancer stem cells to the development of castration resistance of prostate cancer. Mol Cancer. 2014; 13:55.
35. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15–20.
36. Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS biology. 2005; 3:e85.
37. Cesca F, Yabe A, Spencer-Dene B, Scholz-Starke J, Medrihan L, Maden CH, Gerhardt H, Orriss IR, Baldelli P, Al-Qatari M, Koltzenburg M, Adams RH, Benfenati F, et al. Kidins220/ARMS mediates the integration of the neurotrophin and VEGF pathways in the vascular and nervous systems. Cell Death Differ. 2012; 19:194–208.
38. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013; 32:5501–11.
39. Thieu W, Tilki D, deVere White RW, Evans CP. The role of microRNA in castration-resistant prostate cancer. Urol Oncol. 2014; 32:517–23.
40. Knuuttila M, Yatkin E, Kallio J, Savolainen S, Laajala TD, Aittokallio T, Oksala R, Hakkinen M, Keski-Rahkonen P, Auriola S, Poutanen M, Makela S. Castration induces up-regulation of intratumoral androgen biosynthesis and androgen receptor expression in an orthotopic VCaP human prostate cancer xenograft model. Am J Pathol. 2014; 184:2163–73.
41. Selth LA, Townley S, Gillis JL, Ochnik AM, Murti K, Macfarlane RJ, Chi KN, Marshall VR, Tilley WD, Butler LM. Discovery of circulating microRNAs associated with human prostate cancer using a mouse model of disease. Int J Cancer. 2012; 131:652–61.
42. Yaman Agaoglu F, Kovancilar M, Dizdar Y, Darendeliler E, Holdenrieder S, Dalay N, Gezer U. Investigation of miR-21, miR-141, and miR-221 in blood circulation of patients with prostate cancer. Tumour Biol. 2011; 32:583–8.
43. Zhang HL, Yang LF, Zhu Y, Yao XD, Zhang SL, Dai B, Zhu YP, Shen YJ, Shi GH, Ye DW. Serum miRNA-21: elevated levels in patients with metastatic hormone-refractory prostate cancer and potential predictive factor for the efficacy of docetaxel-based chemotherapy. Prostate. 2011; 71:326–31.
44. Sun T, Yang M, Chen S, Balk S, Pomerantz M, Hsieh CL, Brown M, Lee GS, Kantoff PW. The altered expression of MiR-221/-222 and MiR-23b/-27b is associated with the development of human castration resistant prostate cancer. Prostate. 2012; 72:1093–103.
45. Jiao J, Hindoyan A, Wang S, Tran LM, Goldstein AS, Lawson D, Chen D, Li Y, Guo C, Zhang B, Fazli L, GleaveM, Witte ON, et al. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS One. 2012; 7:e42564.
46. Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, Calhoun-Davis T, Zaehres H, Daley GQ, Tang DG. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009; 27:993–1005.
47. Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ, Tang DG. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011; 30:3833–45.
48. Lukacs RU, Memarzadeh S, Wu H, Witte ON. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell. 2010; 7:682–93.
49. Rybak AP, Tang D. SOX2 plays a critical role in EGFR-mediated self-renewal of human prostate cancer stem-like cells. Cell Signal. 2013; 25:2734–42.
50. Kregel S, Kiriluk KJ, Rosen AM, Cai Y, Reyes EE, Otto KB, Tom W, Paner GP, Szmulewitz RZ, Vander Griend DJ. Sox2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PLoS One. 2013; 8:e53701.
51. Rustagi Y, Rani V. Screening of MicroRNA as potential CardiomiRs in Rattus noveregicus Heart related Dataset. Bioinformation. 2013; 9:919–22.
52. van Rooij E, Purcell AL, Levin AA. Developing microRNA therapeutics. Circ Res. 2012; 110:496–507.
53. Iglesias T, Cabrera-Poch N, Mitchell MP, Naven TJ, Rozengurt E, Schiavo G. Identification and cloning of Kidins220, a novel neuronal substrate of protein kinase D. J Biol Chem. 2000; 275:40048–56.
54. Kong H, Boulter J, Weber JL, Lai C, Chao MV. An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J Neurosci. 2001; 21:176–85.
55. Bracale A, Cesca F, Neubrand VE, Newsome TP, Way M, Schiavo G. Kidins220/ARMS is transported by a kinesin-1-based mechanism likely to be involved in neuronal differentiation. Mol Biol Cell. 2007; 18:142–52.
56. Higuero AM, Sanchez-Ruiloba L, Doglio LE, Portillo F, Abad-Rodriguez J, Dotti CG, Iglesias T. Kidins220/ARMS modulates the activity of microtubule-regulating proteins and controls neuronal polarity and development. J Biol Chem. 2010; 285:1343–57.
57. Lopez-Menendez C, Gascon S, Sobrado M, Vidaurre OG, Higuero AM, Rodriguez-Pena A, Iglesias T, Diaz-Guerra M. Kidins220/ARMS downregulation by excitotoxic activation of NMDARs reveals its involvement in neuronal survival and death pathways. J Cell Sci. 2009; 122:3554–65.
58. Neubrand VE, Cesca F, Benfenati F, Schiavo G. Kidins220/ARMS as a functional mediator of multiple receptor signalling pathways. J Cell Sci. 2012; 125(Pt 8):1845–54.
59. Cesca F, Yabe A, Spencer-Dene B, Arrigoni A, Al-Qatari M, Henderson D, Phillips H, Koltzenburg M, Benfenati F, Schiavo G. Kidins220/ARMS is an essential modulator of cardiovascular and nervous system development. Cell Death Dis. 2011; 2:e226.
60. Jean-Mairet RM, Lopez-Menendez C, Sanchez-Ruiloba L, Sacristan S, Rodriguez-Martinez M, Riol-Blanco L, Sanchez-Mateos P, Sanchez-Madrid F, Rodriguez-Fernandez JL, Campanero MR, Iglesias T. The neuronal protein Kidins220/ARMS associates with ICAM-3 and other uropod components and regulates T-cell motility. Eur J Immunol. 2011; 41:1035–46.
61. Liao YH, Hsu SM, Huang PH. ARMS depletion facilitates UV irradiation induced apoptotic cell death in melanoma. Cancer Res. 2007; 67:11547–56.
62. Rogers DA, Schor NF. Kidins220/ARMS is expressed in neuroblastoma tumors and stabilizes neurotrophic signaling in a human neuroblastoma cell line. Pediatr Res. 2013; 74:517–24.
63. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009; 9:550–62.
64. Jiang BH, Liu LZ. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res. 2009; 102:19–65.
65. Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014; 16:378–86.
66. Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocrine-related cancer. 2013; 20:R83–99.
67. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11–22.
68. Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, Wu H. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011; 19:792–804.
69. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer cell. 2011; 19:575–86.
70. Jia S, Gao X, Lee SH, Maira SM, Wu X, Stack EC, Signoretti S, Loda M, Zhao JJ, Roberts TM. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013; 3:44–51.
71. Akiri G, Nahari D, Finkelstein Y, Le SY, Elroy-Stein O, Levi BZ. Regulation of vascular endothelial growth factor (VEGF) expression is mediated by internal initiation of translation and alternative initiation of transcription. Oncogene. 1998; 17:227–36.
72. Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007; 17:71–7.
73. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001; 21:3995–4004.
74. Choi YH, Jin GY, Li LC, Yan GH. Inhibition of protein kinase C delta attenuates allergic airway inflammation through suppression of PI3K/Akt/mTOR/HIF-1 alpha/VEGF pathway. PLoS One. 2013; 8:e81773.
75. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe’er J, Trent JM, Meltzer PS, Hendrix MJ. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999; 155:739–52.
76. Folberg R, Maniotis AJ. Vasculogenic mimicry. APMIS. 2004; 112:508–25.
77. Kluetz PG, Figg WD, Dahut WL. Angiogenesis inhibitors in the treatment of prostate cancer. Expert Opin Pharmacother. 2010; 11:233–47.
78. Conteduca V, Aieta M, Amadori D, De Giorgi U. Neuroendocrine differentiation in prostate cancer: current and emerging therapy strategies. Critical reviews in oncology/hematology. 2014; 92:11–24.
79. Poon M, Zhang X, Dunsky KG, Taubman MB, Harpel PC. Apolipoprotein(a) induces monocyte chemotactic activity in human vascular endothelial cells. Circulation. 1997; 96:2514–9.
80. Hu M, Wang C, Li W, Lu W, Bai Z, Qin D, Yan Q, Zhu J, Krueger BJ, Renne R, Gao SJ, Lu C. A KSHV microRNA Directly Targets G Protein-Coupled Receptor Kinase 2 to Promote the Migration and Invasion of Endothelial Cells by Inducing CXCR2 and Activating AKT Signaling. PLoS Pathog. 2015; 11:e1005171.
81. Aranda E, Owen GI. A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA.hy926 endothelial cell line. Biol Res. 2009; 42:377–89.
82. Mao X, Yu Y, Boyd LK, Ren G, Lin D, Chaplin T, Kudahetti SC, Stankiewicz E, Xue L, Beltran L, Gupta M, Oliver RT, Lemoine NR, et al. Distinct genomic alterations in prostate cancers in Chinese and Western populations suggest alternative pathways of prostate carcinogenesis. Cancer Res. 2010; 70:5207–12.
83. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921.