
INTRODUCTION
Colorectal cancer (CRC) is one of the most common cancers, and CRC-related deaths are highly prevalent worldwide including Korea, which has become a significant public health in most developed countries [1] Moreover, the five-year survival rate remains quite low in advanced or metastatic CRC [2]. According to the annual report of the Korea Central Cancer Registry, a total of 24,986 CRC cases detected in Korea in 2009, representing 13.0% of all incident cancer cases [3, 4]. CRC is the second and third most common cancer in Korean men and women, respectively [3, 4].
The nuclear factor-erythroid 2-related factor 2 (Nrf2) is a transcription factors which are activated in response to cellular stress. Nrf2 binds to ARE in the promoter regions of target genes for antioxidant enzymes [5–7]. Nrf2 is traditionally regarded as the cellular defense mechanism and cell survival [6–8] and has been recently expected as a novel target for the chemoprevention of CRC [8]. Many factors including oxidative stress, reactive nitrogen species (RNS) and reactive oxygen species (ROS) can cause chronic inflammation. It is currently well known that chronic inflammation is a major factor contributing to 15-20% cancers including CRC. However, recent studies have revealed the bad side of Nrf2 which promotes the survival of cancer cells as well as normal cells [9].
HMG-CoA reductase inhibitors (statins) are widely prescribed drugs used in the treatment of dyslipidemia to prevent cardiovascular or cerebrovascular events. Their therapeutic role has been known not only as their cholesterol-lowering effect but also as their pleiotropic actions which means anti-inflammatory, anti-thrombotic, vascular cytoprotection, anti-oxidant, immunomodulatory, or anti-cancer effects of statin [10, 11]. They are also known to increase atherosclerotic plaque stability, to improve endothelial function and to have an anti-oxidant effect and anti-inflammatory activity [12]. According to some studies that investigate the antioxidant and anti-inflammatory effect of statins, atorvastatin activates heme oxygenase-1 at the stress response elements in prostatic cancer cells and breast cancer cells [13]. Simvastatin decreases reactive oxygen species level by Nrf2 activation via PI3K/Akt pathway in primary mouse embryonic fibroblasts from wild-type or Nrf2 knockout C57BL6J mice and ST-2 cells [14]. Simvastatin stimulates HO-1 expression in Neuro 2A cells by Nrf2 protein [15]. Fluvastatin prevents oxidative stress in vascular smooth muscle via the Nrf2-dependent antioxidant pathway [16]. These studies have shown that statin activates HO-1 and decrease reactive oxygen species level and have anti-oxidant effect in some cancer cells, fibroblasts, and neuronal cells. In our previous study, we have also revealed that simvastatin activates the apoptosis of colon cancer cells and inhibits IGF-1 induced ERK and Akt via the downregulation of IGF-1R expression and proapoptotic ERK activation [in press]. However, there are little known how statins affect activation of Nrf2 and HO-1 antioxidant systems in colon cancer cells. In this study, we investigated the effect of simvastatin on the expression of Nrf2 and HO-1 related antioxidants in two colon cancer cell lines, HT-29, and HCT-116.
RESULTS
Simvastatin induces Nrf2 expression and nuclear translocation of Nrf2
The transcription factor Nrf2 is a member of the basic leucine zipper NF-E2 family and interacts with the ARE in the promoter of phase II detoxifying enzymes, including the gene encoding HO-1. Therefore, we examined whether simvastatin could induce Nrf2 activation. As shown in Figure 1A and 1B, treatment with simvastatin increased expression of Nrf2 in a dose-dependent manner which was reversed by mevalonate treatment in HCT116 and HT-29 cells. Next, we examined whether simvastatin could induce nuclear translocation of Nrf2 from the cytosol to the nucleus. As shown in Figure 2A and 2C, treatment with simvastatin increased nuclear accumulation of Nrf2 in a dose-dependent manner. We also examined nuclear translocation of Nrf2 using an immunocytochemistry assay in HCT116 and HT-29. Figure 2B and 2D showed that treatment of 10 μM of simvastatin for 24 h increased nuclear accumulation of Nrf2 protein from the cytoplasm to the nucleus by immunocytochemical analysis.
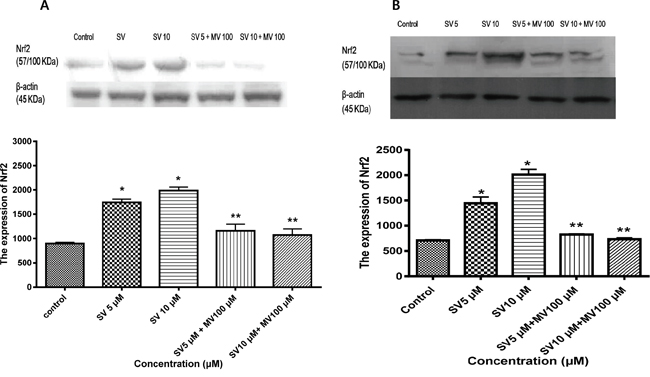
Figure 1: Simvastatin induces Nrf2 expression in HCT116 and HT-29 cells. A. HCT116 cells were incubated with the indicated concentrations of simvastatin for 24 h. Nrf2 expression was determined using Western blotting. Simvastatin increases expression of Nrf2 in a dose-dependent manner (*p < 0.001 compared with control), which is reversed by mevalonate treatment (**p < 0.001 compared with simvastatin alone). SV, simvastatin; MV, mevalonic acid. B. HT-29 cells were incubated with the indicated concentrations of simvastatin for 24 h. Nrf2 expression was determined using Western blotting. The data represent three independent experiments. Simvastatin increases expression of Nrf2 in a dose-dependent manner (*p < 0.001 compared with control), which is reversed by mevalonate treatment (**p < 0.001 compared with simvastatin alone). SV, simvastatin; MV, mevalonic acid.
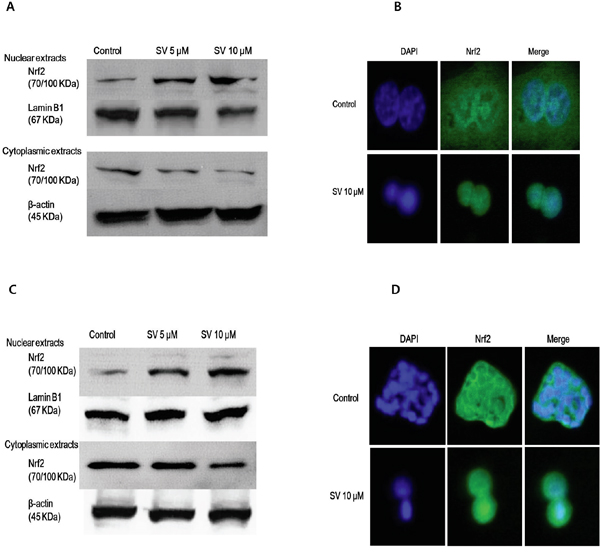
Figure 2: Effect of simvastatin on nuclear translocation of Nrf2. A. HCT116 cells were treated with 5 μM and 10 μM of simvastatin for 24 h. Nuclear fractions and cytoplasmic fractions were respectively analyzed by Western blotting for the detection of Nrf2 nuclear translocation Simvastatin increases the nuclear accumulation of Nrf2 in a dose-dependent manner. B. HCT116 cells were treated with 10 μM simvastatin for 24 h. After treatment, cell was fixed and labeled with an anti-Nrf2 antibody, followed by a fluorescein isothiocyanate (FITC)-conjugated secondary antibody. The nuclei were visualized by DAPI staining. Simvastatin for 24 h increased the nuclear accumulation of Nrf2 protein from the cytoplasm to the nucleus by immunocytochemical analysis. C. HT-29 cells were treated with the indicated concentrations of simvastatin for 24 h. Nuclear fractions and cytoplasmic fractions were respectively analyzed by Western blotting for the detection of Nrf2 nuclear translocation. Simvastatin increased the nuclear accumulation of Nrf2 in a dose-dependent manner. D. HT-29 cells were treated with 10 μM simvastatin for 24 h. After treatment, cells were fixed and labeled with an anti-Nrf2 antibody, followed by an FITC-conjugated secondary antibody. The nuclei were visualized by DAPI staining. Simvastatin for 24 h increased the nuclear accumulation of Nrf2 protein from the cytoplasm to the nucleus by immunocytochemical analysis. The data represent three independent experiments. SV, simvastatin.
Simvastatin induces Nrf2-related antioxidant expression in HCT116 and HT-29 cells
Nrf2 binds to AREs and positively regulates the expression and induction of HO-1 and NAD(P)H: quinine oxidoreductase 1 (NQO1) genes. To investigate the effect of statins on the Nrf2–antioxidant system, we examined three representative genes: HO-1, NQO1, and γ-glutamate-cysteine ligase catalytic subunit (GCLC) by western blotting. To assess the role of simvastatin in the induction of HO-1 expression, HCT116 and HT-29 were treated for 24 h with simvastatin at doses of 5 μM and ten μM and HT-29. After treatment with simvastatin for 24 h, the expression of the HO-1 protein was increased in a concentration-dependent manner and reversed by mevalonate treatment on HCT116 and HT-29 cells (Figure 3A and 3B). We also observed that treatment with simvastatin on HCT116 and HT-29 cells led a significant increase in expression for NQO1 and GCLC with concentration-dependent manner (Figure 3A and 3B). These effects were statistically significant increases compared with the control. Thus, we speculate that the simvastatin upregulated Nrf2-related antioxidant enzymes in HCT116 and HT-29 cells.
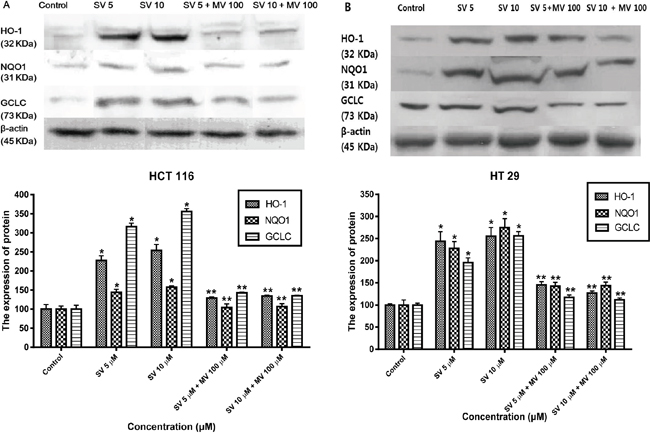
Figure 3: Simvastatin induces transcriptional activation of HO-1, NQO1, and GCLC. A. HCT116 cells were treated with the indicated concentrations of simvastatin for 24 h. The expression of HO-1, NQO1, and GCLC were determined by Western blot analysis. Simvastatin increased the expression of HO-1, NQO1, and GCLC in a dose-dependent manner (*p < 0.001 compared with control), which is reversed by mevalonate treatment (**p < 0.001 compared with simvastatin alone). B. HT-29 cells were treated with the indicated concentrations of simvastatin for 24 h. The expression of HO-1, NQO1, and GCLC were determined by Western blot analysis. Simvastatin increased the expression of HO-1, NQO1, and GCLC in a dose-dependent manner (*p < 0.001 compared with control), which is reversed by mevalonate treatment (**p < 0.001 compared with simvastatin alone). The data represent three independent experiments. SV, simvastatin; MV, mevalonic acid.
Simvastatin activates Nrf2 and HO-1 through ERK and PI3K/Akt pathway
Previous studies reported that mitogen-activated protein kinase (MAPK)/ERK and phosphoinositide 3-kinase (PI3K)/Akt signaling pathways are controlled by reactive oxygen species and are associated with HO-1 expression [17–20]. Therefore, we examined the possibility that the MAPK/ERK and PI3K/Akt pathways are involved in the simvastatin-induced HO-1 expression. First, we investigated whether simvastatin could activate ERK and if those signals were associated with simvastatin-induced HO-1 expression in HCT116 and HT-29. Treatment with simvastatin (5 μM in HCT-116 and ten μM in HT-29) for 24 h increased phosphorylation of ERK, an MAPK inhibitors, 25 μM PD98059 (ERK inhibitor) and mevalonate (100 μM) for two h respectively reduced simvastatin-induced Nrf2 and HO-1 expression (Figure 4A and 4B). These results indicate that ERK signaling is associated with simvastatin-induced HO-1 expression. Next, we investigated whether simvastatin increased HO-1 expression via activation of PI3K/Akt pathway. In the presence of simvastatin (5 μM in HCT-116 and ten μM in HT-29) for 24 h, Akt phosphorylation increased. Pretreatment of cells with ten μM LY294002, a PI3K inhibitor and mevalonate (100 μM) respectively inhibited simvastatin-induced Nrf2 and HO-1 expression (Figure 5A and 5B). Taken together, these results indicate that both simvastatin-induced ERK and PI3K/Akt pathways play a critical role in HO-1 induction via Nrf2 nuclear translocation in HCT116 and HT-29 cells.
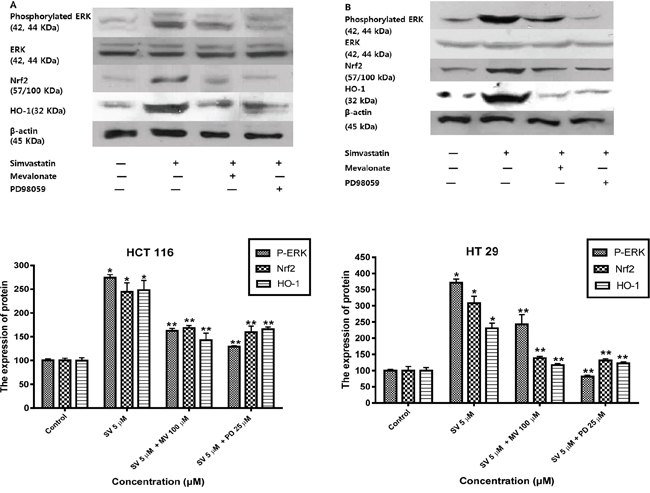
Figure 4: Simvastatin-induced ERK phosphorylation activates Nrf2 and HO-1 expression in HCT116 and HT-29 cells. A. HCT116 cells were pretreated with or without PD98059 (25 μM) or mevalonate (100 μM) for 2h and then co-treated with simvastatin (5 μM) for 24 h. Western blot analysis was performed. Simvastatin increased the expression of phosphorylated ERK, Nrf2, and HO-1 in a dose-dependent manner (*p < 0.001 compared with control), which is reversed by mevalonate or PD98059 treatment (**p < 0.001 compared with simvastatin alone). SV, simvastatin; MV, mevalonic acid; PD, PD98059. B. HT-29 cells were pretreated with or without PD98059 (25 μM) or mevalonate (100 μM) for 2h and then co-treated with simvastatin (10 μM) for 24 h. Western blot analysis was performed. The data represent three independent experiments. Simvastatin increased the expression of phosphorylated ERK, Nrf2, and HO-1 in a dose-dependent manner (*p < 0.001 compared with control), which is reversed by mevalonate or PD98059 treatment (**p < 0.001 compared with simvastatin alone). SV, simvastatin; MV, mevalonic acid; PD, PD98059.
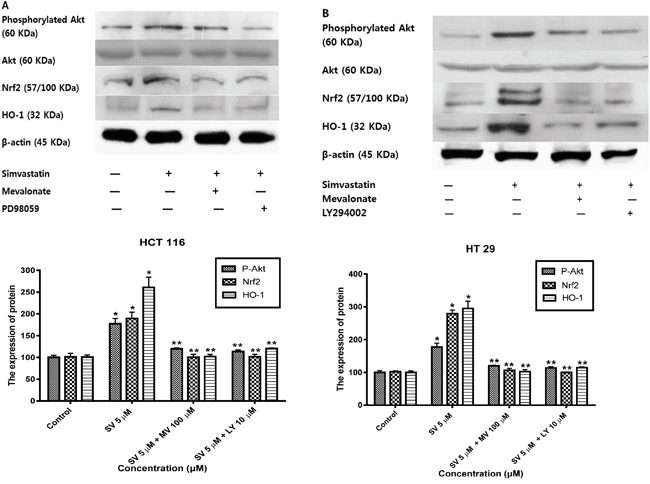
Figure 5: Simvastatin-induced PI3K/Akt phosphorylation activates Nrf2 and HO-1 expression in HCT116 and HT-29 cells. A. HCT116 cells were pretreated with or without LY294002 (10 μM) or mevalonate (100 μM) for 2h and then co-treated with simvastatin (5 μM) for 24 h. Western blot analysis was performed. Simvastatin increased the expression of phosphorylated Akt, Nrf2, and HO-1 in a dose-dependent manner (*p < 0.001 compared with control), which is reversed by mevalonate or LY294002 treatment (**p < 0.001 compared with simvastatin alone). SV, simvastatin; MV, mevalonic acid; LY, LY294002. B. HT-29 cells were pretreated with or without LY294002 (10 μM) or mevalonate (100 μM) for 2h and then co-treated with simvastatin (10 μM) for 24 h. Western blot analysis was performed. The data represent three independent experiments. Simvastatin increased the expression of phosphorylated Akt, Nrf2, and HO-1 in a dose-dependent manner (*p < 0.001 compared with control), which is reversed by mevalonate or LY294002 treatment (**p < 0.001 compared with simvastatin alone). SV, simvastatin; MV, mevalonic acid; LY, LY294002.
DISCUSSION
We had demonstrated that simvastatin inhibited HT-29 cell proliferation and induced apoptosis through the caspase cascade and affecting the expression of the Bcl-2 family proteins in our previous study (in press). The results of our present study demonstrated that simvastatin-induced HO-1 expression by nuclear translocation of Nrf2 via both ERK and PI3K/Akt pathway in HCT116 and HT-29 colon cancer cells.
HO-1 is a key protein that plays a central role in the cellular adaptation to stress induced by many pathological events [21]. Although the key factors participating in signal transduction mechanisms needed for transcriptional activation of HO-1 remain to be fully identified, this enzyme can be regarded as a potential therapeutic target for a variety of oxidant- and inflammatory diseases [22]. Statins induce HO-1 in several cell types, such as vascular smooth muscle cells, endothelial cells, and macrophages. The anti-inflammatory effects of statins have been linked to the induction of the cytoprotective gene encoding HO-1 [23]. Statin is known to activate the HO-1 promoter in endothelial cells and reduce free radical formation. This effect leads to their antioxidant and anti-inflammatory actions [24, 25]. The HO enzyme family degrades heme to biliverdin, thereby providing carbon monoxide (CO) and free iron (Fe2). Both bilirubin (the product of biliverdin degradation) and CO may protect molecules against oxidative stress and ischemia and reperfusion injury [26–29]. They reduce superoxide anions, suppress lipid peroxidation, prevent apoptosis, increase vasodilatation, and improve local blood flow [30, 31]. However, the role of HO-1 in cancer is controversial. HO-1 can be expressed at high levels in some tumor cells, and down-regulation of HO-1 by HO-1-shRNA or inhibition of the enzyme by specific inhibitor has been shown to inhibit proliferation of some hormone-refractory prostate cancer cells [32]. On the other hand, pro-apoptotic and anti-proliferative functions of HO-1 have been reported in prostate cancer [33], breast cancer [34] and oral cancer [35], although the mechanism of action is not known. In a previous study, HO-1 determines the differential response of breast cancer and normal cells to piperlongumine [36]. This study suggested that HO-1 has an anti-tumor function in cancer cells, but cytoprotective functions in normal cells. In our study, simvastatin activated HO-1 expression in colon cancer cells but, it is uncertain whether HO-1 expression by simvastatin protects cancer cells or inhibits cell proliferation in colon cancer cells.
Nrf2 is an important transcription factor that activates expression of antioxidant defense genes through binding to ARE in the promoter region of antioxidant genes such as HO-1 and glutathione regulatory enzymes in response to ROS [37, 38]. The activity of Nrf2 is normally suppressed in the cytosol by Keap1 protein. The treatment of cells with ARE inducers causes dissociation of Nrf2 from Keap1, allowing to Nrf2 translocate to the nucleus. In our study, we found that simvastatin-induced Nrf2 activation and nuclear translocation, which increased the expression of its downstream target enzymes, HO-1, NQO1 and GCLC in HT-29 and HCT116 cells. Nrf2 plays dual roles in cancer prevention and progression depending on the cellular context and environment. Nrf2 is considered a major mechanism of protection against chemical and radiation capable of damaging DNA integrity and initiating carcinogenesis [39]. It protects cells against chemical and radiation stress and promotes cell survival. Simvastatin activates Keap1/Nrf2 signaling in rat liver to protect the cells from the deleterious effects of oxidative stress [40]. Lovastatin protects neural stem cells against oxidative stress-induced cell death via expression of anti-apoptotic Nrf2 and PGC-1α [41]. Fluvastatin has cytoprotective effects against oxidative stress, activating antioxidant genes through Nrf2/ARE in human coronary artery smooth muscle cells [16]. In this study, simvastatin activated Nrf2 nuclear translocation, HO-1 expression, and then the expression of HO-1 related antioxidants in colon cancer cells. However, in this study, it is unclear how simvastatin-induced Nrf2 and HO-1 activation interact with apoptosis stimulated by simvastatin in colon cancer cells. It remains to investigate further study to evaluate the role of HO-1 stimulated by simvastatin in colon cancer.
Modification of the Nrf2–Keap1 complex and Nrf2 nuclear translocation is necessary to Nrf2–ARE-dependent gene expression, and several signaling pathways are associated with processes depending on cell type or stimulators [42]. The MAPKs are a family of signal-transducing enzymes participating in the regulation of many essential cellular processes. Many studies have focused on the resolution of MAPKs pathways in the activation of HO-1 in diverse cell types in response to various inducing conditions [43–46]. ERK and p38MAPK induce Nrf2 translocation and HO-1 expression through diallyl sulfide in HepG2 cells [47]. In human aortic smooth muscle cells, oxidized low-density lipoprotein induces nuclear translocation of Nrf2 via activation of MAPK [48].
A recent study showed that ERK phosphorylates Nrf2, which may induce the release of Nrf2 from the Keap1-Nrf2 complex, causing activated Nrf2 to translocate to the nucleus [49]. In other studies, PI3K/Akt has an important role in Nrf2 signaling. In response to the herb-derived phenol carnosol, Nrf2–ARE binding activity increased. A PI3K inhibitor and dominant negative mutant of Akt blocked carnosol-induced HO-1 expression in PC12 cells [48]. The present experiment designed to determine a possible role of MAPK/ERK pathway and PI3K/Akt pathway in simvastatin-induced HO-1 expression showed that simvastatin activates ERK and Akt phosphorylation. Moreover, this study demonstrated that the expression of Nrf2 and HO-1 are activated via ERK and PI3K/Akt signaling in colon cancer cells.
Taken together, we demonstrated that simvastatin activates Nrf2 activation and nuclear translocation of Nrf2 and then induces the expression of HO-1 related antioxidants via ERK and PI3K/Akt pathway in HT-29 cells. Further studies are needed to explore what the exact role of Nrf2 and HO-1 related antioxidants stimulated by simvastatin is while simvastatin suppresses cell proliferation and increases apoptosis in colon cancer cells.
MATERIALS AND METHODS
Materials
Dulbecco's modified Eagle medium (DMEM), fetal bovine serum (FBS), trypsin/EDTA and penicillin/streptomycin were from Gibco (Grand Island, NY, USA). Simvastatin were from Calbiochem (Gibbstown, NJ, USA), Beta-actin antibody (sc-130656, polyclonal, rabbit, 1:1000), HO-1 antibody (sc-10789, polyclonal, rabbit, 1:1000), Nrf2 antibody (sc-722, polyclonal, rabbit, 1:1000), NQO1 antibody (sc-25591, polyclonal, rabbit, 1:1000), and Goat anti-rabbit IgG-HRP (sc-2004, polyclonal, rabbit, 1:1000), were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), Phopho-p44/42 MAP kinase antibody (9101s, polyclonal, rabbit, 1:1000), non-phospho-p44/42 MAP kinase antibody (9102s, polyclonal, rabbit, 1:1000), Phospho-Akt antibody (9271s, polyclonal, rabbit, 1:1000), non-phospho-Akt antibody (9272s, polyclonal, rabbit, 1:1000) were from Cell signaling (Danvers, MA, USA), GCLC antibody (ab154770, polyclonal, rabbit, 1:300) was from Abcam (Cambridge, MA, USA), and Amersham ECL™ Advance Western Blotting Detection Kit were from Amersham Biosciences (Buckinghamshire, UK).
Cell culture
HT-29 cells were provided by Dr. C.S. Eun (Hanyang University College of Medicine, Seoul, Korea). HCT-116 cells were purchased from Korean Cell Line Bank (KCLB). Cells were cultured in DMEM with two mM glutamine and 4.5 g/L glucose supplemented with 1.5 g/L sodium bicarbonate, 10 % FBS, 100 μg/mL streptomycin, and 100 IU/ml penicillin. We exchanged the media twice a week, and the cells were kept in 37°C incubator with 5 % CO2. We subcultured cells when confluent (every 5~7 days) using EDTA (1 g/L) and trypsin (2.5 g/L). Experiments were carried out serum-free medium (SFM) containing 0.1 % bovine serum albumin (BSA, Sigma, St Louis, MO, USA).
MTT (3-[4,5-dimethylthiazol-2-yl]-diphenyltetrazolium bromide) Assay
We seeded cells at a density of 5X 104 cells/mL with the cultured medium in a 96-well plate. After incubation for 24 h, cells were cultured in various concentration of simvastatinin serum-free medium for 24 or 48 h. Then MTT (0.5 mg/mL) (Sigma, St. Louis, MO, USA) was added to each well and incubated for further four hour 37°C. After the medium has been removed, 100 μL of dimethylsulfoxide (DMSO) was added to each well by shaking the plate for 10 min. The optical density (OD) was evaluated by DTX 880 Multimode Detector (Beckman Coulter, Brea, CA, USA) at 570 nm. Each assay was performed in triplicates.
Western blotting
Cells were washed with PBS and harvested with lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 % Tripton X-100, 1 μM phenylmethylsulfonyl fluoride (PMSF), 1 % sodium deoxycholate, 0.1 % SDS, 5 μg/ml aprotinin, 5 μg/ml leupeptin). Protein contents were analyzed using the Bradford assay (Sigma, St Louis, MO, USA). SDS-PAGE was performed with a 4 % stacking gel and a 10 % resolving gel, followed by transfer to nitrocellulose membrane (Bio-rad, Hercules, CA, USA). We blocked the membranes for one hour at room temperature in blocking solution (5 % skim milk in Tris-buffer with Tween-20 [TBS-T]: 200 mM Tris, 500 mM NaCl, pH 7.5, 0.05 % v/v Tween-20), and then incubated overnight at 4°C in 5 % BSA solution (5% bovine serum albumin in TBS-T) with Nrf2 antibody, HO-1 antibody, NQO1 antibody, GCLC antibody, beta-actin antibody, Phopho-p44/42 MAP kinase antibody, non-phospho-p44/42 MAP kinase antibody, Phospho-Akt antibody, or non-phospho-Akt antibody. The membranes were washed with TBS-T and incubated with Goat anti-rabbit IgG-HRP for one hour at room temperature. The membrane washed and incubated with Amersham ECL™ Advance Western Blotting Detection Kit for 2 min, and autoradiography was checked. The signal intensities of specific bands on the Western blotting were quantified using National Institutes of Health Image J density analysis software (Version1.43).
Nuclear extract preparation
Nuclear extracts for western blotting and immunofluorescence staining were prepared by Nuclear/Cytosol Fractionation Kit (Biovision, Milpitas, CA, USA). Cells were washed with ice-cold PBS and collected by centrifugation at 600 x g for 5 min at 4°. We resuspended cell pellets in 0.2 mL cytosol extraction buffer A containing DTT and protease inhibitors and kept on ice for 10 minutes. The supernatant obtained was added to 11 μL of cytosol extraction buffer B, maintained on ice for 1 min, and centrifuged at 16,000 x g for 5 min. The supernatant (cytosol extract) was transferred to a new tube. The pellet (contains nuclei) was resuspended in 100 μL of nuclear extraction buffer mix, vortexed on ice for 15 seconds, and then centrifuged at 16,000 x g for 10 min. The supernatant (nuclear extract protein) concentration was measured using the Bradford method for western blotting.
Immunofluorescence staining
Cells were cultured on glass coverslips in 6-well plates and treated with simvastatin (10 μM) for 24 h. Cells were fixed in ice cold methanol for 10 min and incubated with a Nrf2 antibody (1:100) (Santa Cruz Biotechnology, United States) in PBS containing 1% BSA for one h. After three washes with PBS, cells were incubated with the FITC-conjugated secondary antibody for one h in PBS containing 1% FBS and washed with PBS for three times. The cells on coverslips were incubated with DAPI (0.5 μg/mL) (Sigma, St. Louis, MO, USA) for 1 min. Images obtained using Olympus IX81 fluorescence microscope (Olympus, USA) and processed using Xcellence 1.2 software (Olympus, USA).
Statistical analysis
Results from each experiment were expressed as the mean±SD of three separate experiments. Data were analyzed One-way analysis of variance (ANOVA) by Tukey's multiple comparisons tests using GraphPad Prism 4.0 software.
ACKNOWLEDGMENTS
This study was supported by a research grant from JW Pharmaceutical (913032).
CONFLICTS OF INTEREST
The authors have no commercial associations that might represent a conflict of interest about this manuscript.
DISCLOSURE
This study was presented in the poster session of United European Gastroenterology Week 2015.
FINANCIAL SUPPORT
This study was supported by a research grant from JW Pharmaceutical (913032).
Author contributions
These authors have all approved the final draft.
Hyun Joo Jang: The conception and design of the study; analysis and interpretation of the data; drafting the article as the first author and corresponding author
Jin Lee: The conception and design of the study; revising it critically for important intellectual content as the co-corresponding author
Eun Mi Hong: The conduction of main experiments
Mikang Kim, Jae Hyun Kim, Juah Jang, Se Woo Park, Hyun Woo Byun, Dong Hee Koh, Min Ho Choi, Sea Hyub Kae: acquisition of data; analysis and interpretation of the data.
Abbreviations
HO-1, heme-oxygenase-1; Nrf2, NF-E2-related factor; ERK, extracellular-signal-regulated kinase activity/protein; ARE, antioxidant responsive element; NQO1, NAD(P)H: quinine oxidoreductase 1; GCLC, γ-glutamate-cysteine ligase catalytic subunit.
REFERENCES
1. Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, Jemal A, Schymura MJ, Lansdorp-Vogelaar I, Seeff LC, van Ballegooijen M, Goede SL, Ries LA. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010; 116:544-573.
2. O'Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004; 96:1420-1425.
3. Jung KW, Park S, Kong HJ, Won YJ, Lee JY, Seo HG, Lee JS. Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2009. Cancer Res Treat. 2012; 44:11-24.
4. Park SH, Song CW, Kim YB, Kim YS, Chun HR, Lee JH, Seol WJ, Yoon HS, Lee MK, Lee JH, Bhang CS, Park JH, Park JY, Do BH, Park YD, Yoon SJ, et al. Clinicopathological characteristics of colon cancer diagnosed at primary health care institutions. Intest Res. 2014; 12:131-138.
5. Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009; 48:91-104.
6. Yu S, Kong AN. Targeting carcinogen metabolism by dietary cancer preventive compounds. Curr Cancer Drug Targets. 2007; 7:416-424.
7. Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008; 52:S128-138.
8. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001; 357:539-545.
9. Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacological research. 2008; 58:262-270.
10. McFarlane SI, Muniyappa R, Francisco R, Sowers JR. Clinical review 145: Pleiotropic effects of statins: lipid reduction and beyond. J Clin Endocrinol Metab. 2002; 87:1451-1458.
11. Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005; 45:89-118.
12. Adam O, Laufs U. Antioxidative effects of statins. Arch Toxicol. 2008; 82:885-892.
13. Kwok SC, Samuel SP, Handal J. Atorvastatin activates heme oxygenase-1 at the stress response elements. J Cell Mol Med. 2012; 16:394-400.
14. Chartoumpekis D, Ziros PG, Psyrogiannis A, Kyriazopoulou V, Papavassiliou AG, Habeos IG. Simvastatin lowers reactive oxygen species level by Nrf2 activation via PI3K/Akt pathway. Biochem Biophys Res Commun. 2010; 396:463-466.
15. Hsieh CH, Rau CS, Hsieh MW, Chen YC, Jeng SF, Lu TH, Chen SS. Simvastatin-induced heme oxygenase-1 increases apoptosis of Neuro 2A cells in response to glucose deprivation. Toxicol Sci. 2008; 101:112-121.
16. Makabe S, Takahashi Y, Watanabe H, Murakami M, Ohba T, Ito H. Fluvastatin protects vascular smooth muscle cells against oxidative stress through the Nrf2-dependent antioxidant pathway. Atherosclerosis. 2010; 213:377-384.
17. Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, De Galarreta CM, Cuadrado A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004; 279:8919-8929.
18. Takeda K, Lin J, Okubo S, Akazawa-Kudoh S, Kajinami K, Kanemitsu S, Tsugawa H, Kanda T, Matsui S, Takekoshi N. Transient glucose deprivation causes upregulation of heme oxygenase-1 and cyclooxygenase-2 expression in cardiac fibroblasts. J Mol Cell Cardiol. 2004; 36:821-830.
19. Papaiahgari S, Zhang Q, Kleeberger SR, Cho HY, Reddy SP. Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxidants & redox signaling. 2006; 8:43-52.
20. Cheng PY, Lee YM, Shih NL, Chen YC, Yen MH. Heme oxygenase-1 contributes to the cytoprotection of alpha-lipoic acid via activation of p44/42 mitogen-activated protein kinase in vascular smooth muscle cells. Free radical biology & medicine. 2006; 40:1313-1322.
21. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A. 1968; 61:748-755.
22. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes & development. 1999; 13:76-86.
23. Lee TS, Chang CC, Zhu Y, Shyy JY. Simvastatin induces heme oxygenase-1: a novel mechanism of vessel protection. Circulation. 2004; 110:1296-1302.
24. Grosser N, Hemmerle A, Berndt G, Erdmann K, Hinkelmann U, Schurger S, Wijayanti N, Immenschuh S, Schroder H. The antioxidant defense protein heme oxygenase 1 is a novel target for statins in endothelial cells. Free radical biology & medicine. 2004; 37:2064-2071.
25. Grosser N, Erdmann K, Hemmerle A, Berndt G, Hinkelmann U, Smith G, Schroder H. Rosuvastatin upregulates the antioxidant defense protein heme oxygenase-1. Biochem Biophys Res Commun. 2004; 325:871-876.
26. Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H, Yoshida K, Nagai R. Carbon monoxide protects against cardiac ischemia--reperfusion injury in vivo via MAPK and Akt--eNOS pathways. Arterioscler Thromb Vasc Biol. 2004; 24:1848-1853.
27. Neto JS, Nakao A, Kimizuka K, Romanosky AJ, Stolz DB, Uchiyama T, Nalesnik MA, Otterbein LE, Murase N. Protection of transplant-induced renal ischemia-reperfusion injury with carbon monoxide. Am J Physiol Renal Physiol. 2004; 287:F979-989.
28. Kawamura K, Ishikawa K, Wada Y, Kimura S, Matsumoto H, Kohro T, Itabe H, Kodama T, Maruyama Y. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler Thromb Vasc Biol. 2005; 25:155-160.
29. Adin CA, Croker BP, Agarwal A. Protective effects of exogenous bilirubin on ischemia-reperfusion injury in the isolated, perfused rat kidney. Am J Physiol Renal Physiol. 2005; 288:F778-784.
30. Sato K, Balla J, Otterbein L, Smith RN, Brouard S, Lin Y, Csizmadia E, Sevigny J, Robson SC, Vercellotti G, Choi AM, Bach FH, Soares MP. Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J Immunol. 2001; 166:4185-4194.
31. Soares MP, Usheva A, Brouard S, Berberat PO, Gunther L, Tobiasch E, Bach FH. Modulation of endothelial cell apoptosis by heme oxygenase-1-derived carbon monoxide. Antioxidants & redox signaling. 2002; 4:321-329.
32. Alaoui-Jamali MA, Bismar TA, Gupta A, Szarek WA, Su J, Song W, Xu Y, Xu B, Liu G, Vlahakis JZ, Roman G, Jiao J, Schipper HM. A novel experimental heme oxygenase-1-targeted therapy for hormone-refractory prostate cancer. Cancer research. 2009; 69:8017-8024.
33. Gueron G, De Siervi A, Ferrando M, Salierno M, De Luca P, Elguero B, Meiss R, Navone N, Vazquez ES. Critical role of endogenous heme oxygenase 1 as a tuner of the invasive potential of prostate cancer cells. Mol Cancer Res. 2009; 7:1745-1755.
34. Hill M, Pereira V, Chauveau C, Zagani R, Remy S, Tesson L, Mazal D, Ubillos L, Brion R, Asghar K, Mashreghi MF, Kotsch K, Moffett J, Doebis C, Seifert M, Boczkowski J, et al. Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: mutual cross inhibition with indoleamine 2,3-dioxygenase. FASEB J. 2005; 19:1957-1968.
35. Lee YM, Jeong GS, Lim HD, An RB, Kim YC, Kim EC. Isoliquiritigenin 2'-methyl ether induces growth inhibition and apoptosis in oral cancer cells via heme oxygenase-1. Toxicol In Vitro. 2010; 24:776-782.
36. Lee HN, Jin HO, Park JA, Kim JH, Kim JY, Kim B, Kim W, Hong SE, Lee YH, Chang YH, Hong SI, Hong YJ, Park IC, Surh YJ, Lee JK. Heme oxygenase-1 determines the differential response of breast cancer and normal cells to piperlongumine. Mol Cells. 2015; 38:327-335.
37. Cuadrado A, Rojo AI. Heme oxygenase-1 as a therapeutic target in neurodegenerative diseases and brain infections. Curr Pharm Des. 2008; 14:429-442.
38. Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003; 43:233-260.
39. Giudice A, Montella M. Activation of the Nrf2-ARE signaling pathway: a promising strategy in cancer prevention. Bioessays. 2006; 28:169-181.
40. Habeos IG, Ziros PG, Chartoumpekis D, Psyrogiannis A, Kyriazopoulou V, Papavassiliou AG. Simvastatin activates Keap1/Nrf2 signaling in rat liver. J Mol Med (Berl). 2008; 86:1279-1285.
41. Abdanipour A, Tiraihi T, Noori-Zadeh A, Majdi A, Gosaili R. Evaluation of lovastatin effects on expression of anti-apoptotic Nrf2 and PGC-1alpha genes in neural stem cells treated with hydrogen peroxide. Mol Neurobiol. 2014; 49:1364-1372.
42. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes & development. 2013; 27:2179-2191.
43. Chen K, Maines MD. Nitric oxide induces heme oxygenase-1 via mitogen-activated protein kinases ERK and p38. Cell Mol Biol (Noisy-le-grand). 2000; 46:609-617.
44. Kietzmann T, Samoylenko A, Immenschuh S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J Biol Chem. 2003; 278:17927-17936.
45. Wu CC, Hsieh CW, Lai PH, Lin JB, Liu YC, Wung BS. Upregulation of endothelial heme oxygenase-1 expression through the activation of the JNK pathway by sublethal concentrations of acrolein. Toxicol Appl Pharmacol. 2006; 214:244-252.
46. Cooper KL, Liu KJ, Hudson LG. Contributions of reactive oxygen species and mitogen-activated protein kinase signaling in arsenite-stimulated hemeoxygenase-1 production. Toxicol Appl Pharmacol. 2007; 218:119-127.
47. Gong P, Hu B, Cederbaum AI. Diallyl sulfide induces heme oxygenase-1 through MAPK pathway. Arch Biochem Biophys. 2004; 432:252-260.
48. Anwar AA, Li FY, Leake DS, Ishii T, Mann GE, Siow RC. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: role of mitogen-activated protein kinases and Nrf2. Free radical biology & medicine. 2005; 39:227-236.
49. Zipper LM, Mulcahy RT. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol Sci. 2003; 73:124-134.