
INTRODUCTION
Cancer of unknown primary (CUP) refers to solid malignancies where no primary tumor is detected despite appropriate staging [1–3]. 50–70% of CUP cases are adenocarcinomas and 20–30% undifferentiated carcinomas. Squamous cell carcinomas, neuroendocrine carcinomas and other rare malignancies together account for the remaining 10% of CUP cases [1–4]. By combining traditional histologic, immunohistochemical and clinical criteria, 10–30% of CUP patients can be assigned to prognostically favorable subgroups, which include patients that benefit from management in analogy to specified organ cancers and cases with locally restricted disease warranting curative approaches [2, 4, 5]. The prognosis of the remaining majority of patients is dismal. The standard approach in these cases is to offer non-specific cytostatic therapy in palliative intention [2, 3, 6].
In an increasing number of tumor entities, drugs designed to target molecular alterations have been successfully implemented into therapeutic strategies. Ideally, these targeted therapies should be applied as guided by molecular predictors, e.g. activating EGFR mutations triggering therapy by drugs targeting EGFR [7–9]. In CUP, targeted therapies are not established, which is explained by the lack of data on molecular alterations in this entity. The majority of publications on mutations and other aberrations of potential oncogenic drivers in CUP date back to the era before high-throughput sequencing and suffer from small sample sizes and heterogeneous methodology [10–12]. Only lately, two studies on next-generation sequencing of CUP samples were published [13, 14].
In the present study, we characterized molecular aberrations in CUP cases belonging to the most relevant histological categories of adeno- or undifferentiated carcinomas using a panel of 50 genes selected according to their relevance as potential therapeutic targets or biologically important oncogenic drivers. Moreover, detailed clinical data including progression-free survival (PFS) and overall survival (OS) were available, thereby enabling in-depth analyses of relevant clinical correlations.
RESULTS
Patient characteristics
A total of 128 CUP patients with adeno- or undifferentiated carcinoma meeting the eligibility criteria were identified. After exclusion of cases due to insufficient tissue or DNA quality, withdrawal of consent, or revision of diagnosis, the final study population consisted of 55 successfully sequenced cases with complete clinical datasets (Supplementary Figure 1). The high dropout rate due to lack of representative tissue material is explained by the fact that in CUP, the histological diagnosis is usually established on core needle biopsies which have to be subjected to extensive immunophenotyping to exclude organ-specific differentiation. The median follow-up of patients was 28.9 months. During follow-up, 39 deaths and 38 disease progressions were observed. OS was 7 months for male patients and 17 months for female patients (P = 0.14). Patient characteristics are summarized in Table 1.
Table 1: Patient characteristics (n = 55)
Age at diagnosis |
59 ± 10 years* |
|
|---|---|---|
Gender |
||
Females |
28 cases |
(51%) |
Males |
27 cases |
(49%) |
Affected organ sites |
||
Lymph nodes |
33 cases |
(60%) |
Liver |
17 cases |
(31%) |
Bones |
14 cases |
(25%) |
Lung |
11 cases |
(20%) |
Peritoneum |
10 cases |
(18%) |
Adrenals |
8 cases |
(15%) |
Brain |
2 cases |
(4%) |
Skin |
2 cases |
(4%) |
Others organ sites |
7 cases |
(13%) |
Number of affected organ sites |
||
1 |
24 cases |
(44%) |
2 |
15 cases |
(28%) |
3 |
11 cases |
(20%) |
4 |
2 cases |
(4%) |
5 |
2 cases |
(4%) |
Mean |
1.9 ± 1.1* |
|
Median progression-free survival |
7.0 (5.0–9.0) months† |
|
Median overall survival |
16.2 (7.6–23.4) months† |
|
Annotations: *, mean ± standard deviation. †, median with 95% confidence interval.
Molecular alterations in CUP patients
Panel sequencing of 50 selected genes (Supplementary Table 1) revealed at least one molecular alteration in 46 out of 55 cases (84%), including mutations in 43 cases (78%) and CNVs (amplifications/deletions), as revealed by coverage analysis, in 29 cases (53%). In total, 60 tumor-specific mutations and 29 CNVs were detected (Supplementary Table 2). Individual cases harbored up to 5 mutated genes, 4 amplified genes, 2 deleted genes, or 6 molecular alterations in total (Figure 1). The gene most frequently affected was TP53, of which 33 mutations were detected in 30 cases (55%), including three cases with two mutations per case. CDKN2A was affected in 12 cases (22%) including 8 deletions (15%) and 5 mutations (9%). One case showed a CDKN2A deletion together with a CDKN2A mutation. KRAS was affected in 10 cases (18%) including 9 mutations (16%) and one amplification (2%). The 2 detected EGFR mutations were located within exons 18 and 21, compatible with activating mutations [9], and 2 out of 3 BRAF mutations coded for the V600E mutant protein [15]. In both instances, these are established molecular predictors for approved targeted therapies in some entities.
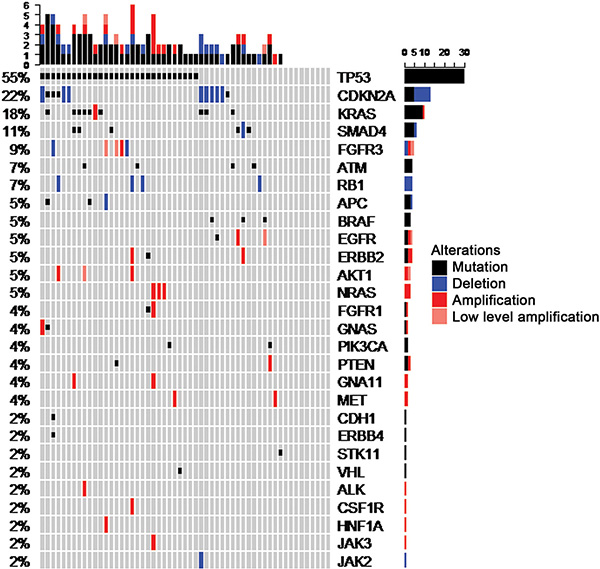
Figure 1: OncoPrint summarizing mutations and copy number alterations of the 55 CUP cases included in the final dataset.
Correlation of molecular alterations to clinical data
The mutational status of the most frequently altered genes, TP53, CDKN2A, KRAS and SMAD4, was tested for correlations to survival data (Table 2). Mutations of KRAS and CDKN2A significantly correlated to poor PFS, with KRAS mutations also showing a significant correlation to poor OS. TP53 mutations were associated with a slightly shorter OS, but this association was not significant based on the log-rank test. Applying a test also able to detect crossing hazard rates [16] revealed a P value of 0.055, indicating the possibility that TP53 mutations may confer an increased risk of early death. Moreover, the mutational status of TP53 interacted with the prognostic impact of gender (Supplementary Figure 2): A significantly better OS (P = 0.003) and PFS (P = 0.007) of female versus male patients was only found in cases lacking TP53 mutations (PFS interaction P =0.08, OS interaction P = 0.27).
Table 2: Correlations of frequent molecular alterations to survival data
Molecular alteration |
Overall survival* |
Progression–free survival* |
|
|---|---|---|---|
TP53 mutation |
present (n= 30) |
10.7 (6.0–NA) months |
7.0 (4.1–13.0) months |
absent (n = 25) |
17.4 (7.6–27.0) months |
7.4 (4.2–12.1) months |
|
P† |
0.85 |
0.97 |
|
KRAS mutation |
present (n= 9) |
6.0 (4.9–NA) months |
3.3 (2.0–NA) months |
absent (n = 46) |
17.4 (8.7–25.4) months |
7.4 (5.9–12.1) months |
|
P† |
0.016 |
0.005 |
|
CDKN2A deletion |
present (n= 8) |
15.4 (7.1–NA) months |
7.9 (3.0–NA) months |
absent (n = 47) |
16.2(7.6–23.8) months |
6.8 (5.0–9.0) months |
|
P† |
0.71 |
0.56 |
|
CDKN2A mutation |
present (n= 5) |
5.9 (3.3–NA) months |
2.9 (1.0–NA) months |
absent (n = 50) |
16.2 (7.6–23.8) months |
7.4 (5.7–10.0) months |
|
P† |
0.16 |
0.015 |
|
SMAD4 mutation |
present (n= 5) |
13.0 (8.2–NA) months |
8.5 (5.9–NA) months |
absent (n = 50) |
16.2 (7.4–25.4) months |
6.8 (4.1–9.0) months |
|
P† |
0.65 |
0.90 |
|
Annotations: *, median with 95% confidence interval. †, log-rank test.
As compared to samples with wildtype TP53, TP53 mutations were associated with a significantly higher number of additional molecular alterations (Table 3). Cases with TP53 mutations were significantly younger than cases without this alteration (56.4 ± 11.3 versus 62.8 ± 6.9 years, P = 0.04). All five cases aged younger than 45 years and 9 out of 10 cases aged 50 years or younger displayed a TP53 mutation. Molecular CDKN2A alterations were associated with lung metastases: 5 out of 11 cases (46%) with altered CDKN2A but only 6 out of 43 cases (14%) with wild-type CDKN2A showed lung involvement (P = 0.03).
Table 3: Number of additional molecular alterations in relation to TP53 mutational status
TP53 mutated (30 cases) |
TP53 unmutated (25 cases) |
P* |
|||
|---|---|---|---|---|---|
Number of additional mutations (in addition to TP53) |
0.96 |
||||
0 |
13 cases |
(43%) |
12 cases |
(48%) |
|
1 |
10 cases |
(33%) |
10 cases |
(40%) |
|
2 |
5 cases |
(17%) |
3 cases |
(12%) |
|
3–4 |
2 cases |
(7%) |
0 cases |
(0%) |
|
Mean |
0.9 ± 1.0 |
0.6 ± 0.7 |
|||
Number of deletions |
0.55 |
||||
0 |
21 cases |
(70%) |
18 cases |
(72%) |
|
1–2 |
9 cases |
(30%) |
7 cases |
(28%) |
|
Mean |
0.3 ± 0.5 |
0.3 ± 0.6 |
|||
Number of amplifications |
0.12 |
||||
0 |
18 cases |
(60%) |
21 cases |
(84%) |
|
1 |
10 cases |
(33%) |
4 cases |
(16%) |
|
2–4 |
2 cases |
(7%) |
0 cases |
(0%) |
|
Mean |
0.7 ± 1.0 |
0.2 ± 0.4 |
|||
Total number of molecular alterations (in addition to TP53) |
0.03 |
||||
0 |
3 cases |
(10%) |
9 cases |
(36%) |
|
1 |
14 cases |
(47%) |
7 cases |
(28%) |
|
2 |
4 cases |
(13%) |
5 cases |
(20%) |
|
3 |
3 cases |
(10%) |
4 cases |
(16%) |
|
4–5 |
6 cases |
(20%) |
0 cases |
(0%) |
|
Mean |
1.9 ± 1.4 |
1.2 ± 1.1 |
|||
Annotation: *, Fisher’s exact test.
DISCUSSION
This study comprises a collection of 55 clinically annotated CUP cases with adeno- or undifferentiated carcinoma. By restricting the inclusion criteria to these histologic categories, representing roughly 90% of all CUP patients, we sought to avoid our results to be confounded by the inclusion of rare CUP subgroups. Although no confirmatory conclusions can be expected from this exploratory investigation, our results are suitable to generate hypotheses that might be subject to confirmatory investigations.
The most frequent molecular alterations affected TP53, CDKN2A and KRAS, which were also the top-three alterations in the only other large study of CUP cases [14]. The frequency of TP53 mutations resembles their frequency in cancer in general [17]. It should be noted that the data on TP53 alterations in CUP published before the advent of next-generation sequencing were heterogeneous and inconclusive, which is likely explained by small sample sizes and the heterogeneity of both methods and inclusion criteria [10–12].
Overall, the molecular heterogeneity of our cohort does not support the assumption of common biological mechanisms underlying the formation of CUP, instead favoring the notion that CUP is a heterogeneous group of different molecular and clinical entities. A noteworthy correlation was the association of TP53 mutations with higher numbers of additional molecular alterations, which fits well to the role of TP53 in maintaining genomic integrity [18, 19], and the correlation of TP53 mutations with genomic alterations known from other entities, e. g. complex aberrant karyotypes in acute myeloid leukemia [20]. In several tumor entities, TP53 mutations have been proposed as predictors of poor prognosis [20–22]. In our study on CUP, however, the prognostic impact of TP53 mutations was less clear, which might be owing to the fact that CUP is a prognostically dismal disease in general and small prognostic differences conferred by TP53 mutations might have evaded detection. Furthermore, since cancer-associated TP53 mutations comprise a heterogeneous spectrum of functional defects [23–25], individual mutations might differ from each other with regard to their prognostic impact, and patient populations with different spectra of TP53 mutations might differ with regard to the role of these mutations as prognostic markers. Interestingly, women not harboring TP53 mutations constituted a subgroup with relatively favorable prognosis. One might speculate that within this subgroup, gynecological cancers are enriched. Indeed, some gynecological cancers, e.g. cervical cancer, are characterized by relatively low percentages of TP53-mutant cases [17], and in several types of gynecological cancers, e.g breast and ovarian cancer, TP53 mutations are known to confer a poor prognosis [22, 26, 27].
Another interesting finding is the relatively high frequency of CDKN2A alterations, being 22% in our cohort and 19% in another study on CUP cases [14]. Frequent CDKN2A deletions have been described in some human cancers [28, 29] while the overall frequency of mutations in CDKN2A in human cancer has been found to be only 3.8% [17]. It should be mentioned that among the major categories of human tumors, the highest rate of CDKN2A mutations has been found in pancreatic cancer [17, 30], and that the mutational spectrum of pancreatic cancer is comparable to the overall spectrum of mutations in CUP detected by us and others [14, 17, 30]. This may either indicate a general biological similarity between these two entities or a frequent origin of CUP from the pancreas, an association that is supported by autopsy studies but weakened by more recent gene expression profiling analyses, which suggest a pancreatic origin of CUP in only 5–12% of cases [31, 32].
The only gene whose mutation was correlated to OS in our cohort was KRAS. As for CDKN2A, pancreatic cancer comprises the highest percentage of cases with mutant KRAS [17]. In addition, KRAS mutations have been linked to poor prognosis in this entity [33], again hinting at a possible similarity between pancreatic cancer and CUP. It should be noted that pharmacologic inhibition of the RAS pathway is among the major goals of current anti-cancer drug development, however, drugging mutant RAS itself is not yet feasible [34].
One might define an alteration as druggable when two conditions are met: Firstly, an approved drug has to be available, and secondly, an alteration must be established as a molecular predictor with regard to this drug. Assuming that data on molecular predictors can be transferred from other entities to CUP, 6 of our cases (11%) harbored druggable alterations: 2 cases with BRAF V600E mutations, 2 cases with activating EGFR mutations, and 2 cases with amplification of ERBB2. One might expand this list by the cases with MET or EGFR amplification, since approved drugs targeting the respective gene products are available. This would elevate the number of potentially druggable patients to 8 cases (15%). Considering the limited therapeutic benefit from currently used standard cytostatic regimens, we conclude that a significant minority of CUP patients may benefit from molecularly stratified therapies. It should be noted that in other defined large entities with comparable frequencies of druggable mutations such as lung cancer, broad spectrum upfront molecular testing is already clinical routine, arguing for comprehensive routine testing in CUP as well. The proportion of druggable cases may rise in the future since drugs targeting additional drivers may soon become available. Examples include FGFR1/3, with specific drugs in late development [35], and the RAS pathway, as already discussed. Clinical trials assessing such molecularly stratified approaches for CUP are urgently needed. In addition, molecular alterations may be useful both as prognostic and predictive markers, e.g. KRAS mutations indicating a poor prognosis and, at the same time, predicting lack of response to therapies targeting EGFR, since it is well established that their action requires an intact downstream RAS pathway [34, 36].
We conclude that the vast majority of CUP tumors harbor mutations of relevant driver genes. At least a significant minority of CUP patients are candidates for molecularly stratified therapies, which may contribute to improve the prognosis of this devastating disease.
MATERIALS AND METHODS
Patients
Patients were eligible if either adenocarcinoma or undifferentiated carcinoma was histologically confirmed by a board-certified pathologist, and if a primary lesion was not detected despite appropriate search including, as a minimum requirement, cross-sectional imaging of chest and abdomen. 36 cases (Heidelberg cohort) were outpatients seen at the National Center for Tumor Diseases (NCT), Heidelberg, Germany. 19 cases (PACET-CUP cohort) were participants of a German multi-center trial (PACET-CUP study) conducted by the Arbeitsgemeinschaft Internistische Onkologie, Deutsche Krebsgesellschaft (German Cancer Society).
DNA preparation
After completion of all necessary routine diagnostics, remaining formalin-fixed, paraffin-embedded biopsy specimens were tested for tumor cell content. A tumor cell content of less than 20% and a biopsy size of less than 0.1 cm were considered insufficient for sequencing. Tumor areas were marked on an H&E stained slide. Corresponding tissue areas were microdissected from three subsequent unstained slides. Extraction of genomic DNA was performed after proteinase K digestion and automated purification using the Maxwell 16 Research System (Promega, Madison, USA). DNA content was measured fluorimetrically using the QuBit 2.0 HS DNA Assay (Thermo Fisher Scientific). DNA sequencing grade quality was confirmed using a real-time qPCR-based method (RNAseP Detection system, Thermo Fisher Scientific) [37].
Library preparation and semiconductor sequencing
For library preparation, the multiplex PCR-based Ion Torrent AmpliSeq™ technology (Thermo Fischer Scientific) with the Cancer Hotspot Panel v2 (CHPv2) was used. Amplicon library preparation was performed with the Ion AmpliSeq Library Kit v2.0 using 10 ng of DNA determined by qPCR assay. Briefly, the DNA was mixed with the primer pool, containing all primers for generating the 207 amplicons, and the AmpliSeq HiFi Master Mix in a 20 μl reaction volume and transferred to a PCR cycler (Biometra, Göttingen, Germany). After the end of the PCR, primer end sequences were partially digested using FuPa reagent, followed by ligation of barcoded sequencing adapters (Ion Xpress Barcode Adapters 1–96, Thermo Fisher Scientific). The final library was purified using AMPure XP magnetic beads (Beckman Coulter, Krefeld, Germany) and quantified using qPCR (Ion Library Quantitation Kit, Thermo Fischer Scientific) on a StepOnePlus qPCR machine (Thermo Fischer Scientific). Individual libraries were diluted to a final concentration of 100 pM and eight to ten libraries were pooled and processed to library amplification on Ion Spheres using Ion PGM™ Template OT2 200 Kit. Unenriched libraries were quality-controlled using Ion Sphere quality control measurement on a QuBit instrument. After library enrichment (Ion OneTouch ES), the library was processed for sequencing using the Ion Torrent 200bp sequencing v2 chemistry and the barcoded libraries were loaded onto a 318v2 chip.
Variant calling and annotation
Raw sequencing data were processed using the Ion Torrent Suite Software (version 4.4.3). After base calling, the reads were aligned against the human genome (hg19) using the TMAP algorithm implemented in the Torrent Suite. Variant calling was performed with the variant caller plugin (version 4.4.3) within the Torrent Suite Software using a corresponding bed-file containing the coordinates of the amplified regions. Variant annotation was performed using a custom build variant annotation pipeline in the CLC Genomics Workbench (version 8.0.2). Annotations included information about nucleotide and amino acid changes of RefSeq annotated genes, COSMIC and dbSNP entries as well as detection of possible splice site mutations. For visualization of sequencing and fusion reads, the Integrative Genomic Browser (IGV, http://www.broadinstitute.org/igv/) was used. Only variants with an allele frequency > 5% and minimum coverage > 200 reads were taken into account. For further analysis, only non-synonymous nucleotide exchanges were considered. Each identified variant was compared to entries in the COSMIC, dbSNP and 6500 Exomes databases.
Copy number variations
Copy number variations (CNVs; amplifications and deletions) were identified by using the coverage data summary for each sample and each amplicon generated by the TorrentSuite software. Detection of CNVs was performed according to Endris et al. [37].
Statistical analysis
OS and PFS were calculated from the date of histologic confirmation of diagnosis (Heidelberg cohort) or the date of entry into the PACET-CUP study (PACET-CUP cohort). No relevant differences between these two landmark dates are expected because the PACET-CUP study is a trial of first-line therapy. Distribution of survival times was estimated by the method of Kaplan and Meier. The log-rank test was used to test for differences between groups. Cox regression was used to assess prognostic interaction. A two-stage testing procedure starting with the log-rank test [16] was applied to test for differences in the presence of potentially crossing hazards. Median follow-up time was estimated based on time to censoring [38]. Fisher’s exact test was used to compare distribution of metastases and alterations between groups. Mann-Whitney test was used to compare age distribution between groups. All p-values were two-sided. P values below 0.05 were considered statistically significant. Analyses were performed with statistical software R including add-on packages ComplexHeatmap and TSHRC. If not indicated otherwise, results are summarized as mean ± SD.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the Arbeitsgemeinschaft Internistische Onkologie, Deutsche Krebsgesellschaft (German Cancer Society) and the German Cancer Consortium (DKTK). Further, we acknowledge excellent technical assistance by Kathrin Ridinger, Katja Lorenz and Jennifer Gruber.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Massard C, Loriot Y, Fizazi K. Carcinomas of an unknown primary origin-diagnosis and treatment. Nat Rev Clin Oncol. 2011; 8:701–710.
2. Neben K, Hübner G, Folprecht G, Jäger D, Krämer A. Metastases in the Absence of a Primary Tumor: Advances in the Diagnosis and Treatment of CUP Syndrome. Dtsch Ärztebl Int. 2008; 105:733–740.
3. Varadhachary GR, Raber MN. Carcinoma of unknown primary site. N Engl J Med. 2014; 371: 2040.
4. Löffler H, Puthenparambil J, Hielscher T, Neben K, Krämer A. Patients with cancer of unknown primary: a retrospective analysis of 223 patients with adenocarcinoma or undifferentiated carcinoma. Dtsch Arztebl Int. 2014; 111:481–487.
5. Hainsworth JD, Fizazi K. Treatment for patients with unknown primary cancer and favorable prognostic factors. Semin Oncol. 2009; 36:44–51.
6. Lee J, Hahn S, Kim DW, Kim J, Kang SN, Rha SY, Lee KB, Kang JH, Park BJ. Evaluation of survival benefits by platinums and taxanes for an unfavourable subset of carcinoma of unknown primary: a systematic review and meta-analysis. Br J Cancer. 2013; 108:39–48.
7. Gazdar AF. Personalized medicine and inhibition of EGFR signaling in lung cancer. N Engl J Med. 2009; 361:1018–1020.
8. Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010; 363:301–304.
9. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–1500.
10. Kamposioras K, Pentheroudakis G, Pavlidis N. Exploring the biology of cancer of unknown primary: breakthroughs and drawbacks. Eur J Clin Invest. 2013; 43:491–500.
11. Löffler H, Krämer A. Biologic Features of CUP. In Krämer A, Löffler H (eds): Cancer of Unknown Primary. Cham: Springer International Publishing Switzerland. 2016; 27–44.
12. van de Wouw AJ, Jansen RL, Speel EJ, Hillen HF. The unknown biology of the unknown primary tumour: a literature review. Ann Oncol. 2003; 14:191–196.
13. Tothill RW, Li J, Mileshkin L, Doig K, Siganakis T, Cowin P, Fellowes A, Semple T, Fox S, Byron K, Kowalczyk A, Thomas D, Schofield P et al. Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J Pathol. 2013; 231:413–423.
14. Ross JS, Wang K, Gay L, Otto GA, White E, Iwanik K, Palmer G, Yelensky R, Lipson DM, Chmielecki J, Erlich RL, Rankin AN, Ali SM et al. Comprehensive Genomic Profiling of Carcinoma of Unknown Primary Site: New Routes to Targeted Therapies. JAMA Oncol. 2015; 1:40–49.
15. Cantwell-Dorris ER, O'Leary JJ, Sheils OM. BRAFV600E: implications for carcinogenesis and molecular therapy. Mol Cancer Ther. 2011; 10:385–394.
16. Qiu P, Sheng J. A two-stage procedure for comparing hazard rate functions. Journal of the Royal Statistical Society: Series B (Statistical Methodology). 2008; 70:191–208.
17. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015; 349: 1483–1489.
18. Van Dyke T. p53 and tumor suppression. N Engl J Med. 2007; 356:79–81.
19. Sharpless NE, DePinho RA. p53: good cop/bad cop. Cell. 2002; 110:9–12.
20. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015; 373:1136–1152.
21. Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, Bühler A, Edelmann J, Bergmann M, Hopfinger G, Hensel M, Hallek M, Döhner H et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010; 28:4473–4479.
22. Pharoah PD, Day NE, Caldas C. Somatic mutations in the p53 gene and prognosis in breast cancer: a meta-analysis. Br J Cancer. 1999; 80:1968–1973.
23. Soussi T, Beroud C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer. 2001; 1:233–240.
24. Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene. 1999; 18:477–485.
25. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007; 28:622–629.
26. Sjogren S, Inganas M, Norberg T, Lindgren A, Nordgren H, Holmberg L, Bergh J. The p53 gene in breast cancer: prognostic value of complementary DNA sequencing versus immunohistochemistry. J Natl Cancer Inst. 1996; 88: 173–182.
27. Reles A, Wen WH, Schmider A, Gee C, Runnebaum IB, Kilian U, Jones LA, El-Naggar A, Minguillon C, Schönborn I, Reich O, Kreienberg R, Lichtenegger W et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res. 2001; 7:2984–2997.
28. Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006; 127:265–275.
29. Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, Mao L, Herath J, Jenkins R, Westra W, Rutter JL, Buckler A, Gabrielson E et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet. 1995; 11:210–212.
30. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008; 321:1801–1806.
31. Pentheroudakis G, Golfinopoulos V, Pavlidis N. Switching benchmarks in cancer of unknown primary: from autopsy to microarray. Eur J Cancer. 2007; 43:2026–2036.
32. Hainsworth JD, Rubin MS, Spigel DR, Boccia RV, Raby S, Quinn R, Greco FA. Molecular gene expression profiling to predict the tissue of origin and direct site-specific therapy in patients with carcinoma of unknown primary site: a prospective trial of the Sarah Cannon research institute. J Clin Oncol. 2013; 31:217–223.
33. Bournet B, Buscail C, Muscari F, Cordelier P, Buscail L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur J Cancer. 2016; 54:75–83.
34. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014; 13:828–851.
35. Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer. 2013; 13:772–787.
36. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008; 358:1160–1174.
37. Endris V, Penzel R, Warth A, Muckenhuber A, Schirmacher P, Stenzinger A, Weichert W. Molecular diagnostic profiling of lung cancer specimens with a semiconductor-based massive parallel sequencing approach: feasibility, costs, and performance compared with conventional sequencing. J Mol Diagn. 2013; 15:765–775.
38. Korn EL. Censoring distributions as a measure of follow-up in survival analysis. Stat Med. 1986; 5:255–260.