
INTRODUCTION
One of the main advances in understanding of cancer has been the observation that genetic changes to genes encoding for the epigenetic machinery are common and recurring events in oncogenesis and tumor progression. The application of next-generation sequencing technologies to tumor samples has allowed the identification of novel mutations and structural variations in proteins involved in DNA methylation and post-translational histone modifications, which are dynamically connected each other in the regulation of gene expression. Disruption of this complex epigenetic control mechanism has been frequently described in hematological malignancies, suggesting that alterations in epigenetic regulators may impair the expression of genes that regulate hematopoietic stem cells (HSC) proliferation, survival and stemness [1-4] (Figure 1).
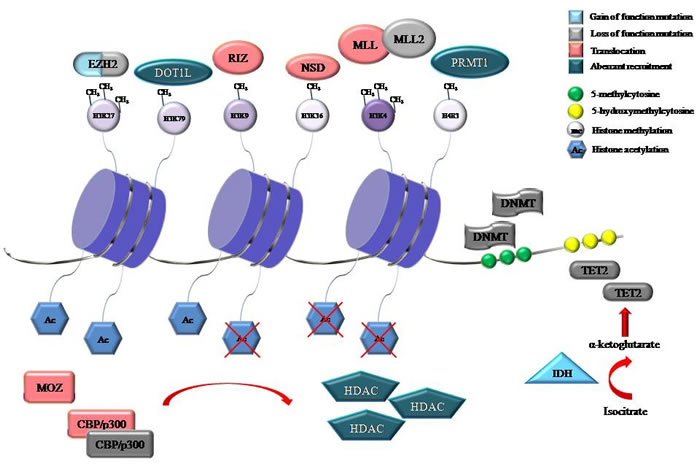
Figure 1: Representative proteins involved in DNA methylation and histone modifications that were identified to be recurrently mutated, translocated or aberrantly recruited in hematological malignancies. The consequences of mutations, either gain of function or loss of function are also shown. Gene symbol: CBP, cyclic AMP response element-binding protein; DNMT, DNA methyltransferase; DOT1L, disruptor of telomeric silencing 1-like; EZH2, enhancer of zeste 2; H3K4, histone 3 lysine 4; H3K9, histone 3 lysine 9; H3K27, histone 3 lysine 27; H3K36, histone 3 lysine 36; H3K79, histone 3 lysine 79; H4R3, histone 4 arginine 3; HDAC, histone deacetylase; IDH, isocitrate dehydrogenase; MLL, mixed-lineage leukemia; MOZ, monocytic leukemia zinc finger protein; NSD, nuclear-receptor-binding SET-domain-containing; PRMT, protein arginine N-methyltransferase; RIZ, retinoblastoma protein-interacting zinc finger; TET2, Ten-Eleven-Translocation 2.
In this review, we mainly focus on epigenetic alterations occurring in hematological malignancies and discuss the potential utility of epigenetic targeting in the treatment of these tumors.
DNA METHYLATION
The best characterized epigenetic modification is DNA methylation, which occurs in the context of cytosine bases in the cytosine-guanine dinucleotides (CpG) in mammalian DNA strands. Cytosine modification is catalyzed by DNA methyltransferases (DNMTs), which transfer a methyl group from the donor S-adenosyl methionine (SAM) to the 5-carbon (C5) position of the pyrimidine ring. Transcriptional silencing by DNA promoter methylation has an essential regulatory function in several cellular processes, and is involved in establishing developmental tissue-specific patterns of gene expression. DNA methylation also plays a crucial role in establishing genomic imprinting and in maintaining balanced expression of X-linked genes in female cells through X chromosome inactivation. DNA methylation also safeguards genomic integrity and stability by silencing endogenous retroviral and parasitic repetitive sequences [5, 6]. Although five members of the DNMT family have been identified, only DNMT1, DNMT3A, and DNMT3B are known to be involved in the methylation of CpG sites. DNMT1 is classified as a maintenance DNMTs since it preferentially copies DNA methylation patterns from a hemi-methylated substrate after DNA replication. Conversely, DNMT3A and DNMT3B serve as de novo DNMTs that are essential in the generation of new methylation patterns during embryogenesis and germ-cell development. Additionally, there is a class of methyl-CpG-binding proteins (MBDs) which bind to methyl-CpG within gene promoters and prevent the binding of specific transcription factors to their recognition sites [7, 8].
Another family of important mediators of DNA methylation is the Ten-Eleven-Translocation (TET) family, which includes proteins that utilize oxygen and α-ketoglutarate (α-KG) to catalyze different reactions, including the oxidation of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC). The TET family of proteins was first identified as a fusion partner of mixed-lineage leukemia (MLL) in patients with t(10;11)(q22;q23) acute myeloid leukemia (AML). Although the biological functions of 5-hmC are still largely unknown, recent evidence suggests that it may play a functional role in stem cell biology [8].
DNA methylation appears to be critically involved in hematopoietic cell differentiation and the development of hematological malignancies, since several genes that regulate the processing of 5-mC are commonly found to be mutated in hematopoietic tumors [9].
DNMTs
DNMTs are essential for early stage of hematopoiesis. The absence of DNMT-1 in HSC leads to impaired self-renewal in vitro. Furthermore, mouse HSC lacking Dnmt-1 were unable to suppress key myeloerythroid regulators and lost their ability to differentiate into lymphoid progeny, thus demonstrating that DNA methylation is necessary to protect normal HSC from lineage restriction [10]. More recently, Challen GA et al. demonstrated that Dnmt3a loss progressively impairs the differentiation capacity of HSC and is accompanied by a simultaneous expansion of HSC in the bone marrow. Furthermore, Dnmt3a-deficient HSC showed both hypermethylation and hypomethylation of promoter regions, resulting in an up-regulation of multipotency genes and a down-regulation of HSC-specific genes. Altogether, these data support the critical involvement of DNMT3A in regulating HSC differentiation [11] and suggest that the loss of de novo DNMT3A activity might impair the differentiation potential of HSC, providing a possible explanation for how DNMT3A mutations can contribute to AML and myelodysplastic syndrome (MDS) pathogenesis. In fact, several studies using large-scale array-based genomic resequencing and whole-genome sequencing of human leukemia have revealed recurrent DNMT3A mutations at multiple sites in AML patients. Greater than 50% of DNMT3A mutations occur at a single amino acid position, R882, located within the catalytic domain; this leads to reduced enzymatic activity in vitro [12]. Consistently, the presence of DNMT3A gene mutations was detected in approximately 20% of patients with AML, a genetic change associated with a shorter overall survival [13]. However, DNMT3A mutations did not correlate with any variations in 5-mC content in AML genomes and were not associated with a specific methylation or gene expression signature in AML patients, so further evaluation is needed to better define the potential pathogenic role of these mutations [13, 14].
DNA hypermethylation
Several lines of evidence point to a role for DNA hypermethylation in the molecular pathogenesis of hematological malignancies (for review see [15]). In fact, the gene encoding the cell cycle regulator p15/INK4b is frequently inactivated by promoter hypermethylation in a large proportion of leukemia patients. Aberrant DNA hypermethylation impairs p15 growth-suppressive properties, allowing leukemic cells to escape inhibitory signals in the bone marrow. Hypermethylation of p15 promoter occurs in approximately 50% of patients with chronic myeloid leukemia (CML), AML, and acute lymphoblastic leukemia (ALL) and represents a key feature of the malignant progression of MDS [16]. In fact, increased CpG methylation at the INK4b locus was associated with the progression of MDS to AML, thus suggesting that aberrant p15 gene hypermethylation may be considered an early event in myeloid cell transformation [17]. A strict association between aberrant promoter methylation and DNMT expression has been found in MDS, a hematological malignancy in which the list of genes inactivated by hypermethylation has grown considerably (for review see [18]). Recently, using an in vitro MDS model, DNA hypermethylation of several genes involved in normal hematopoiesis was identified and associated with elevated DNMT isoform expression, supporting the notion that this disease is characterized by widespread epigenetic deregulation [19].
DNA hypomethylation
Loss of methylation has been reported in several hematological malignancies. Genome-wide DNA methylation takes place predominantly at repetitive sequences, including short and long interspersed nuclear elements and LTR elements, segmental duplications and centromeric and subtelomeric regions [20, 21]. The Long Interspersed Nucleotide Element-1 (LINE-1) repetitive elements are the most well-documented interspersed repetitive elements displaying hypomethylation in various cancers, including ALL. Hypomethylation in the promoter region of LINE-1 can lead to the reactivation of transposable LINE-1 elements that may cause chromosomal instability, as observed in CML [22].
TET enzymes and DNA hydroxymethylation
Mutations in TET2 have been found in a range of hematological malignancies, including AML, MDS, myeloproliferative neoplasms (MPN), and chronic myelomonocytic leukemia (CMML) with frequencies of 24%, 19%, 12% and 22%, respectively [23]. In a study involving patients with MDS and CMML, a TET2 loss-of-function mutation was detected in CD34+ cells, suggesting the early occurrence of this genetic change along the natural history of these malignancies [24]. It has been also reported that loss of Tet2 expression in Tet2-deficient mouse embryonic stem cells led to impaired hematopoietic differentiation, with expansion of HSC and multipotent progenitor cells. Particularly, Tet2-deficient mice developed hematopoietic malignancies resembling human CMML [25]. Altogether, these data might support the critical role for Tet2 in regulating 5-hmC levels within genes involved in the self-renewal, proliferation, and differentiation of HSC. As described above, TET2 function requires α-KG, a substrate produced from isocitrate by isocitrate dehydrogenase (IDH). Mutations affecting IDH1/2 were detected in up to 20 % of AML and consist of single amino acid substitutions occurring within the active site of the enzyme at one of three highly conserved arginine residues. All reported IDH1 and IDH2 mutations promote a new enzyme activity, with the acquisition of the ability to convert α-KG to produce 2-hydroxyglutarate (2-HG), which is similar in structure to α-KG, outcompetes α-KG for binding and inhibiting TET2 enzymes, thus altering gene expression and impairing lineage specific differentiation. Mutations of IDH1/2 were found to be mutually exclusive with TET2 mutations in a large cohort of AML patients. Consistent with a common role in AML pathogenesis, AML samples with mutations in either IDH1/2 or TET2 displayed overlapping DNA methylation signatures, characterized by global promoter hypermethylation [26].
HISTONE MODIFICATION IN HEMATOLOGICAL MALIGNANCIES
Histones are abundant nuclear proteins involved in the formation of nucleosomes, structures upon which eukaryotic DNA is wrapped. Histones H2A, H2B, H3 and H4 can undergo several post-translational modifications mainly in their N-terminal tails, with methylation and acetylation being the most studied. These modifications, in turn, alter the electrostatic charge of histones and modify their binding to DNA, thus playing an essential role in determining the transcriptional status of chromatin, either inducing an open chromatin structure that is permissive for transcription or a more condensed chromatin state, which leads to transcriptional gene silencing. Modifications to histone tails can also create binding surfaces for protein recognition modules, such as bromodomains and chromodomains, which recruit specific functional complexes. So far, a wide range of possible combinations of histone modifications has been identified. Each histone tail can be either unmodified or modified, with lysine residues that can either be mono-, di-, or tri-methylated or acetylated. Based on this intricate pattern of histone modifications, Struhl and Allis proposed a “histone code” hypothesis, suggesting that non-histone proteins would be able to “write”, “read” and “erase” histone modifications and/or their combinations in order to regulate specific chromatin functions and, therefore, to determine the transcriptional status of the target gene (reviewed in [27]). In all higher eukaryotes, canonical histones are encoded by multiple gene copies, giving rise to several variants with different sequences [28]. The “canonical” histones are expressed at high levels during the S-phase of the cell cycle, allowing for their rapid deposition behind the DNA replication fork. By contrast, replication-independent histone “variants” are expressed and incorporated into chromatin throughout the cell cycle (for reviews see [29]). Canonical histone genes are located within multigene clusters, and their expression is largely regulated by a unique 3’ end mRNA which is not polyadenylated, but instead contains a 26 bp sequence that forms a stem-loop structure. On the contrary, genes for histone variants are typically found in a single or low copy number, and are regulated similarly to normal genes [30]. The incorporation of histone variant into nucleosomes may be influenced by DNA methylation. A handful of studies in both plants and mammals report that the H2A variant H2A.Z is excluded from methylated DNA at promoters and within gene bodies, suggesting that epigenetic factors also contribute to histone localization [31, 32].
Mutations affecting genes encoding for canonical histones and their variants have an important role in altering chromatin architecture, leading to aberrant gene expression. Mutations in H3F3A, which encodes the histone H3 variant H3.3, have been recognized as being especially important in promoting aberrant global histone modification changes in pediatric glioblastoma. However, H3F3A mutations represent a very rare event in hematopoietic tumors, since they occur only in a ALL with a very low incidence [33]. In addition to H3F3A, the H2AFX gene also seems to be involved in both cancer initiation and progression. Deletion of band 11q23, where H2AFX maps, has been detected in several human cancers, including T cell prolymphocytic leukemia and B-CLL [34]. Furthermore, a G/A single nucleotide polymorphisms upstream of the start codon of H2AFX has been associated with non-Hodgkin lymphoma (NHL) susceptibility and development [35]. Histone gene mutations are not restricted to histone H2 and H3, since mutations affecting H1 family members have also been observed in follicular lymphoma [36]. H1 gene mutations are located within the H1 globular domain and result in reduced capacity to associate with chromatin [37] and binding to DNMT3B [38]. However, it is not yet clear if histone H1 mutations lead to defective chromatin compaction or result in chromosomal instability.
Histone methylation
Histone methylation involves the addition of methyl groups to the side chain nitrogen atoms of arginine, lysine and histidines residues, with the most commonly histone methylation sites including histone H3 lysine 4 (H3K4), H3K9, H3K27, H3K36, H3K79 and H4K20. Depending on which residue is modified, histone methylation is associated with both transcriptional gene activation and repression. The pattern of histone methylation is read by histone methyltransferases (HMTs) that catalyze the addition of methyl groups donated from S-adenosylmethionine to histones. So far, three families of HMTs have been identified: the SET-domain-containing proteins and disruptor of telomeric silencing 1-like (DOT1L) proteins, which methylate lysines, and members of the protein arginine N-methyltransferase (PRMT) family, which methylate arginines (reviewed in [39]).
The EZ homolog 2 (EZH2) protein contains a SET domain that is responsible for the trimethylation of H3K27, a histone modification associated with transcriptional silencing [40].The EZH2 gene is highly expressed in germinal center B (GCB) cells, indicating it has a role in normal GCB cellular physiology [41]. As B cells exit the GCB, EZH2 expression usually decreases, allowing the expression of genes that are involved in terminal differentiation [42]. A recurrent mutation of tyrosine 641 (Y641) within the EZH2 SET domain has been detected in approximately 22% of diffuse large B-cell lymphoma (DLBCL) and 7% of follicular lymphomas (FL) [43]. This mutation was the first of chromatin-modifying gene alteration to be described in DLBCL and FL. Surprisingly, lymphoma cell lines carrying the Y641 mutation showed increased H3K27 trimethylation. In light of these observations, in vitro experiments demonstrated that the mutated EZH2 form was defective in catalyzing H3K27 monomethylation, but acquired enhanced catalytic efficiency for the H3K27 trimethylation [44, 45]. A global increase in H3K27 trimethylation, due to EZH2 Y641 mutation, represses genes involved in proliferation checkpoints and associated with B-cell differentiation, thus providing a possible explanation for the oncogenic role of mutant EZH2 in DLBCL [41, 46]. A series of loss-of-function EZH2 mutations have been identified in patients with myeloid malignancies, such as MDS, CMML, and primary myelofibrosis [47-49]. Although these studies demonstrated that EZH2 loss resulted in a global decrease in H3K27 trimethylation, the molecular mechanism by which the inactivation of EZH2 contributes to hematopoietic transformation is yet to be determined.
The nuclear-receptor-binding SET-domain-containing (NSD) family consists of three members, NSD1, NSD2, and NSD3, which preferentially target H3K36 methylation. Although NSD proteins are frequently found to be translocated in leukemia, little is known about their role in transcriptional regulation. In childhood AML, the NSD1 gene is disrupted by the t(5,11)(q35;p15.5) chromosomal translocation, which produces the fusion protein NUP98–NSD1 [50]. In vitro studies have indicated that the chimeric protein NUP98-NSD1 promotes the transcription of the Homeodomain (HOX) genes [51], which are normally involved in proliferation and differentiation of HSC. This epigenetic deregulation of HOX genes sustains the self-renewal of myeloid stem cells that is essential for the development of AML. A second gene of the family, NSD2, may fuse with the IgH locus via t(4;14) translocation, which affects 15-20% of patients with multiple myeloma (MM) [52]. Chromosomal fusion leads to overexpression of the NSD2 protein, which may promote the proliferation of MM cells presumably by reprogramming global histone methylation and gene expression [53].
MLL is a SET domain family protein that plays a role in H3K4 methylation. The first gene of this family, MLL, was shown to target SET domain HMT activity to positively regulate HOX genes transcription in hematopoiesis through di- and tri-methylation of H3K4 at HOX promoter and enhancer sequences [54]. Rearrangements affecting the MLL gene are normally found in approximately 10% of AMLs in adults and in more than 70% of infant leukemia [46]. Chromosomal rearrangements involving MLL can occur in several forms such as balanced translocations, inversions, and partial tandem duplications of 11q23. Balanced translocations are the most common rearrangement disrupting MLL and more than 50 different partner genes are known to date [55-57]. Although some of these translocations delete the C-terminal containing the SET domain, and therefore HMT activity [58], the resultant MLL fusion proteins seem to act as dominant positive transcriptional regulators capable of transforming both HSC and early myeloid progenitors. Recent evidence indicates that MLL fusion proteins may be involved in the regulation of gene expression by directing histone modification. The most common MLL fusion proteins, including MLL-AF4, MLL-AF9, and MLL-ENL, belong to a set of multiprotein complexes involved in transcriptional activation/elongation. These complexes can recruit DOT1L, an HMT that lacks the canonical SET domain and is responsible for catalyzing the methylation of H3K79. Studies on different human MLL-rearranged leukemia cells have demonstrated high levels of H3K79 methylation at HOX genes and other MLL targets, thus suggesting that aberrant H3K79 methylation may represent a mechanism of oncogenic transcriptional activation in MLL leukemia [46, 59]. MLL fusion proteins can also transform primary myeloid progenitors by directing the histone arginine methyltransferase PRMT1 to the promoter regions of HOX genes, which become aberrantly methylated at H4 arginine 3 and actively transcribed [60]. Translocations involving MLL has been widely described in the last years, but loss of function mutation affecting MLL2 have also been found in a significant percentage of malignancies, including NHL. In particular, in 89% of FL and 32% of DLBCL, MLL2 mutations resulted in small deletions creating frameshifts or point mutations that introduced premature stop codons in the catalytically active SET domain [43, 61, 62]. However, the effect of MLL2 mutations on H3K4 methylation and the mechanism by which MLL2 loss contributes to lymphomagenesis are still unclear. Another member of this family, MLL3, has been found mutated in FL and DLBCL [63, 64], suggesting a functional redundancy in oncogenic transformation.
The retinoblastoma protein-interacting zinc finger (RIZ) is a HMT involved in H3K9 methylation [65], which is associated with transcriptional silencing. RIZ proteins contain a domain, called the PR domain, showing similarity with the SET domain. The RIZ gene produces two related proteins, RIZ1 and RIZ2, which differ at the N-terminal domain by the presence or absence of the PR domain, respectively. The RIZ1 product commonly undergoes deletions in several types of human cancers. Consistently, mice lacking RIZ1, but not RIZ2, were found to develop unusual tumors, such as diffuse large B-cell lymphoma, suggesting that the PR domain is likely responsible for the tumor suppressing activity of RIZ1 [66]. Another member of the of the RIZ family is the MDS1-EVI1 gene, which maps to the 3q26 locus and encodes two products with different lengths: MSD1-EVI1, containing the PR domain with HMT activity, and EVI1, lacking the PR domain [67]. The PR domain-containing product is known to possess tumor suppressor functions, while EVI1 has a positive role in oncogenesis. MSD1-EVI1 and EVI1 are expressed in hematopoietic cells and their inappropriate expression has been implicated in the development of myeloid disorders. Rearrangements such as translocations or inversions involving the 3q26 band usually cause disruption of the MDS1-EVI1 protein and overexpression of EVI1, as observed in AML and MDS [68, 69]. However, the mechanism by which EVI1 overexpression contributes to oncogenic transformation remains unclear.
Histone acetylation
Acetylation of the ε-amino group of lysine residues is an important post-translational modification that occurs not only in histone tails, but also in chromatin proteins and non-histone proteins regulating important pathways. Acetylation of lysine residues neutralizes the charge on histones, leading to an open chromatin structure that is more permissive for gene transcription, whereas deacetylation of lysines promotes chromatin condensation and transcriptional gene silencing. The dynamic equilibrium of lysine acetylation in vivo depends on the balance of the activities of histone acetyl transferases (HATs) and HDACs [70].
HATs
According to their homology, known HATs can be grouped into different families, including the MYST superfamily, the GCN5 N-acetyltransferase (GNAT) family, and the cyclic AMP response element-binding protein (CBP)/p300 superfamily [70].
MOZ, a member of the MYST family, has been found to acetylate both itself and lysine (K) residues on H2B, H3 and H4 in vitro and H3K9 in vivo. Besides its intrinsic HAT activity, MOZ functions as co-activator of several transcription factors that are involved in the proliferation and differentiation of HSC [71]. Recurrent reciprocal translocations fuse the MOZ gene to genes encoding CBP and p300 or the protein transcription intermediary factor 2 (TIF2). Fusion proteins resulting from MOZ chromosomal translocation can transform hematopoietic progenitors in vitro and induce myeloproliferative disease in vivo [72]. The chimeric protein MOZ-TIF2, which derives from the inversion inv8(p11q13), retains the conserved HAT domain of MOZ and the CBP-interaction domain of TIF2. In one of the earliest studies, Deguchi et al. reported that the expression of MOZ-TIF2 immortalized myeloid progenitors in vitro and induced AML in mice [73]. Further studies in AML demonstrated that MOZ-TIF2 reduced CBP activity in vivo, resulting in aberrant acetylation patterns that inhibited transcriptional activities of critical cell cycle regulators, such as p53 [74].
Similarly to MOZ, CBP is also involved in HSC self-renewal by regulating the activity of transcriptional complexes. Heterozygous loss of CBP impairs HSC self-renewal and predisposes to hematologic malignancies in humans and in mouse models [75]. Consistently, it has been shown that CBP plays an important role in tumorigenesis through its ability to modulate the transcriptional activity of several transcription factors, such as NF-κB and p53 [76]. The CBP gene is located on chromosome 16p13, which is target of translocations in AML and MDS, including MOZ-CBP [t(8;16)(p11;p13)]. This fusion gene encodes for a protein that retains most of the interaction domains of both parental proteins as well as the HAT domain of MOZ. The resulting MOZ-CBP fusion protein has been found to interact with the p65 subunit of NF-κB and to increase the expression of NF-κB target promoters in HSC from individuals with AML [77]. Besides the translocations mentioned above, inactivating mutations and deletions in the HAT coding domain of CBP and its paralog p300 have been detected in approximately 41% of FL and 39% of DLBCL. The majority of these mutations affected only one allele, suggesting that reduced HAT activity may have a critical role in lymphomagenesis. In fact, CBP/p300 genetic lesions impair their ability to acetylate known substrates such as BCL6 and p53, that are themselves targeted by somatic mutation in DLBCL [62]. Reduced p53 tumor suppressor function and constitutive activation of BCL6 oncoprotein may represent alternative oncogenic mechanisms by which CBP/p300 mutations contribute to DLBCL transformation.
HDACs
So far, mammalian HDACs have been divided into four major classes, based on their sequence similarities to yeast HDACs, subunit localizations, and enzymatic activities: class I (HDAC 1, 2, 3, and 8), class II (HDAC 4, 5, 6, 7, 9, and 10), class III or sirtuins (SIRT 1-7) and class IV which consists of HDAC 11 [78]. Class I HDACs, which share homology with the RPD3 yeast protein, are primarily localized in the nucleus, and are ubiquitously expressed in several human cell lines and tissues. Class II HDACs are homologues to the Hda1 yeast protein and are expressed in a limited number of cell types. They are mainly cytoplasmic, but also shuttle between the nucleus and cytoplasm [79]. Sirtuin members, which are homologues to the Sir2 yeast protein, have been found in a wide variety of subcellular locations [80]. Class IV HDACs share sequence similarity with the catalytic core of both Class I and II enzymes and are found predominantly in the cytoplasm where they preferentially deacetylate non-histone proteins [81].
The first evidence of the involvment of HDACs in oncogenesis was derived from hematological malignancies, where fusion proteins resulting from chromosomal translocations were shown to aberrantly recruit HDACs to specific gene promoters, thereby impairing the differentiation and proliferation of myeloid cells [82]. In this respect, acute promyelocytic leukemia (APL) represents a well described model, in which the retinoic acid (RA) pathway is disrupted and the myeloid differentiation is arrested at the promyelocytic stage [83]. RA receptor (RAR)-α is important for myeloid differentiation and acts as a transcriptional regulator by binding its partner RXR. The heterodimerization process enhances RAR-α binding affinity to RA response elements (RAREs) within the promoters of target genes, thus increasing its transcriptional efficiency [81]. In the absence of RA, the RAR-α-RXR heterodimer recruits HDAC-containing repression complexes that lead to chromatin condensation and transcription repression. Physiological concentrations of RA induce a conformational change in the RAR-α-RXR complex, resulting in the dissociation of repression complexes and the subsequent recruitment of transcription factors [84, 85]. In 100% of APL cases, the RAR-α gene is involved in reciprocal chromosomal translocations and, in >90% of these cases, the translocation partner is promyelocytic leukemia protein (PML), resulting in the formation of the PML-RAR-α chimeric protein, which acts as a transcriptional repressor. The PML-RAR-α fusion protein shows an increased ability to bind to DNA at RAREs and to associate with corepressor complexes containing HDACs and chromatin-modifying factors, such as HMTs and DNMTs that mediate the establishment of a repressive chromatin structure at RARα target promoters [86, 87]. PML-RAR-α fusion proteins do not dissociate from corepressor complexes at physiological concentrations of RA, resulting in the transcriptional silencing of RAR-α target genes which normally play an important role in the control of myeloid cell differentiation. Furthermore, PML-RAR-α fusion proteins have been shown to bind to non-canonical RAREs, resulting in widespread transcriptional deregulation that strongly interferes with gene expression programs involved in myeloid proliferation and hematopoietic progenitor self-renewal [88, 89].
Similar mechanisms of transcriptional disruption via histone deacetylation have been described in AML, where the chimeric AML1/ETO gene is produced by the fusion of the AML1 gene on chromosome 21 to the Eight-Twenty One (ETO) oncogene on chromosome 8. The t(8;21) was the first translocation to be discovered and represents one of the most frequent chromosomal abnormalities in AML [90]. Although AML1 functions as a transcriptional activator through the interaction with a complex containing p300, in the AML1-ETO fusion protein the HAT-interacting region of AML1 is replaced by the coding sequence of ETO. The resulting aberrant fusion protein has multiple effects on the regulation of proliferation, differentiation, and viability of leukemic cells by recruiting enzymes involved in epigenetic regulation. In fact, the ETO transcription factor interacts with repressor complex containing HDAC, but also associates with DNMT1, thus imposing aberrant transcriptional repression through intensive histone deacetylation and DNA hypermethylation at AML1 target hematopoietic promoters [88].
Bromodomain proteins
Bromodomains (BRDs) represent highly evolutionarily conserved protein-interaction modules that recognize acetylated lysine residues in histones and other non-histone proteins. To date, over 40 nuclear or cytoplasmic BDRs have been identified, with each protein containing as many as six bromodomains. Human BRDs belong to eight distinct families and include chromatin-modifying enzymes, chromatin remodellers, transcriptional co-activators and mediators, and the large group of bromodomain and extra-terminal motif (BET) proteins. BET proteins utilize dual, tandem N-terminal BRD modules (BD1 and BD2) able to bind to acetylated lysine residues preferentially on H3 and H4 histone tails and on non-histone targets such as the NF-κB subunit RelA and GATA1 [91, 92]. The BET family consists of four members that regulate transcription by RNA polymerase II (Pol II): BRD2, BRD3, BRD4, which are ubiquitously expressed, and BRDT, the expression of which is restricted to germ cells. BET proteins do not possess enzymatic activity at chromatin, but function as scaffolds for a number of transcription factors or chromatin-modifying enzymes (for reviews see [93, 94]). Novel global discovery proteomics have recently identified BET protein as obligate components of core transcriptional regulatory machineries, including the polymerase-associated factor complex (PAFc) and the super elongation complex (SEC), which are critical regulator of MLL translocated leukemias [95]. Furthermore, BET proteins have been found to associate with a great number of active promoters and to converge upon a significant fraction of active enhancers of key genes, including critical oncogenic targets such as c-MYC [96-98]. This suggests a role for BET proteins as epigenetic readers and modifiers in leukemias and other hematologic malignancies.
EPIGENETIC THERAPIES IN HEMATOLOGICAL MALIGNANCIES
As described above, enzymes that maintain and modify the epigenome play a crucial role in the regulation of normal hematopoiesis and, as such, they are often targeted by somatic alterations in hematological malignancies. In this context, epigenetic drugs are considered as an important therapeutic modality for the clinical management of hematological malignancies. Epigenetic drugs can be used alone or in combination therapies with currently available cancer chemotherapies.
DNMT inhibitors
So far, the most widely studied DNMT inhibitors (DNMTi) 5-azacytidine (azacitidine, Vidaza) and 5-aza-2’-deoxycytidine (5-AZA-CdR, Decitabine, Dacogen) have undergone intensive clinical development that led to their Food and Drug Administration (FDA) approval for patients affected by MDS [99]. Once taken up by the cell, the nucleoside analogs azacitidine and 5-AZA-CdR undergo a series of metabolic modifications prior to being incorporated into genomic DNA during the S phase of the cell cycle. After incorporation, they behave as suicide substrates for the DNMTs that, in the attempt to methylate them, became irreversibly inactivated through the formation of a stable covalent bond between the enzyme and the modified pyrimidine ring. This binding leads to depletion of DNMT1 and passive hypomethylation of the genome following DNA replication. Azacitidine incorporates into both RNA and DNA, thus inhibiting protein synthesis, while 5-AZA-CdR is incorporated only into DNA [100]. Azacitidine and 5-AZA-CdR have been employed to treat hematological malignancies for a long time using different doses and schedules, alone or in combination with inhibitors of HDAC (for reviews see [101, 102]) (Table 1).
Table 1: Summary of clinical trials examining DNA hypomethylating agents (DHA) alone or in combination with Histone Deacetylase inhibitors in hematological malignancies
DHA |
Phase |
Combination |
Hematological disease |
Identifier |
State |
Azacitidine |
I |
Entinostat |
AML a, CMML, MDS, |
NCT00101179b |
Ongoing |
MDS,AML,CMML |
NCT00528983 |
Ongoing |
|||
VPA, ATRA |
AML,MDS |
NCT01575691 |
Completed |
||
AML, MDS |
NCT00350818 |
Completed |
|||
Belinostat |
AHM |
NCT00351975 |
Completed |
||
MDS |
NCT01152346 |
Completed |
|||
AML, CMML, MDS |
NCT01519011 |
Completed |
|||
Pracinostat |
AHM, MDS |
NCT00741234 |
Completed |
||
MDS |
NCT01571648 |
Completed |
|||
Panobinostat |
AML, MDS, CMML |
NCT01613976 |
Completed |
||
Sodium phenylbutyrate |
AML, MDS |
NCT00004871 |
Completed |
||
AML, MDS, CMML, HL, MM, NHL |
NCT01908387 |
Terminated |
|||
I/II |
Mocetinostat |
MDS |
NCT02018926 |
Recruiting |
|
Romidepsin |
HL, NHL |
NCT01998035 |
Recruiting |
||
Vorinostat |
AML,DMS |
NCT00392353 |
Ongoing |
||
MDS |
NCT01305460 |
Ongoing |
|||
AML, MDS |
NCT01835587 |
Ongoing |
|||
Vorinostat, Gemtuzumab ogamicin |
AML |
NCT00895934 |
Completed |
||
Mocetinostat |
AML, MDS |
NCT00324220 |
Completed |
||
II |
MDS, JMML |
NCT02447666 |
Recruiting |
||
MDS |
NCT01652781 |
Recruiting |
|||
AML,MDS |
NCT02204020 |
Recruiting |
|||
CMML, MDS |
NCT01404741 |
Recruiting |
|||
AML |
NCT02450877 |
Recruiting |
|||
Vorinostat |
AML, MDS |
NCT01617226 |
Recruiting |
||
Panobinostat |
AML, CMML, MDS |
NCT00946647 |
Recruiting |
||
VPA, Lenalidomide |
MDS |
NCT01342692 |
Recruiting |
||
VPA |
AML, MDS |
NCT02124174 |
Recruiting |
||
AML, MDS |
NCT01995578 |
Recruiting |
|||
AML, MDS |
NCT01462578 |
Recruiting |
|||
MDS |
NCT02281084 |
Recruiting |
|||
Vorinostat |
AML,MDS |
NCT00948064 |
Ongoing |
||
Entinostat |
AML, CMML, MDS, |
NCT00313586 |
Ongoing |
||
MDS |
NCT01599325 |
Ongoing |
|||
Pracinostat |
MDS |
NCT01873703 |
Ongoing |
||
MDS |
NCT00721214 |
Ongoing |
|||
VPA, ATRA |
AML,MDS |
NCT00326170 |
Completed |
||
VPA, Cytarabine |
AML,DMS |
NCT00382590 |
Completed |
||
MDS |
NCT00897130 |
Completed |
|||
AML |
NCT00739388 |
Completed |
|||
AML |
NCT00387647 |
Completed |
|||
MDS |
NCT00384956 |
Completed |
|||
VPA, ATRA |
MDS, AML |
NCT00339196 |
Completed |
||
Sodium phenylbutyrate |
AML, MDS, MM, NHL |
NCT00006019 |
Completed |
||
VPA, ATRA |
MDS |
NCT00439673 |
Completed |
||
MDS |
NCT00102687 |
Completed |
|||
AML, MDS |
NCT00915785 |
Completed |
|||
CMML |
NCT01235117 |
Completed |
|||
MDS |
NCT00446303 |
Terminated |
|||
CLL |
NCT00413478 |
Terminated |
|||
Mocetinostat |
AML,MDS |
NCT00666497 |
Terminated |
||
Mocetinostat |
HL, NHL |
NCT00543582 |
Terminated |
||
II/III |
Lenalidomide, Vorinostat |
CMML, MDS |
NCT01522976 |
Ongoing |
|
III |
AML,MDS |
NCT00887068 |
Recruiting |
||
CCR |
AML |
NCT01074047 |
Ongoing |
||
MDS |
NCT01186939 |
Completed |
|||
MDS |
NCT00071799 |
Completed |
|||
AML, MDS |
NCT00422890 |
Completed |
|||
IV |
MDS |
NCT01201811 |
Completed |
||
Decitabine |
I |
OSU-HDAC42 |
AML |
NCT01798901 |
Ongoing |
Vorinostat |
AML, CMML, MDS |
NCT00357708 |
Completed |
||
Vorinostat |
AML, ALL,CLL, NHL |
NCT00275080 |
Completed |
||
VPA |
NHL |
NCT00109824 |
Completed |
||
VPA |
AML, CLL, SLL |
NCT00079378 |
Completed |
||
AML, ALL |
NCT00042796 |
Terminated |
|||
Romidepsin |
MDS |
NCT00114257 |
Completed |
||
Cytarabine, Vorinostat |
AML |
NCT01130506 |
Completed |
||
MDS |
NCT00941109 |
Completed |
|||
MDS |
NCT00796003 |
Completed |
|||
ALL |
NCT00349596 |
Completed |
|||
AML, MDS |
NCT00049582 |
Terminated |
|||
I/II |
Panobinostat |
AML, MDS |
NCT00691938 |
Ongoing |
|
MDS |
NCT01165996 |
Completed |
|||
MDS, |
NCT00075010 |
Completed |
|||
II |
AML, MDS |
NCT01687400 |
Recruiting |
||
VPA, ATRA |
AML |
NCT00867672 |
Recruiting |
||
VPA |
AML, MDS |
NCT00414310 |
Completed |
||
CMML, MDS |
NCT00067808 |
Completed |
|||
MDS |
NCT00003361 |
Completed |
|||
AML |
NCT00866073 |
Completed |
|||
MDS |
NCT00619099 |
Completed |
|||
CML |
NCT00042003 |
Completed |
|||
CML |
NCT00042016 |
Completed |
|||
CML |
NCT00041990 |
Completed |
|||
MDS |
NCT00744757 |
Completed |
|||
CMML, MDS |
NCT00113321 |
Terminated |
|||
MDS |
NCT01333449 |
Terminated |
|||
III |
MDS |
NCT01751867 |
Completed |
||
MDS |
NCT00043381 |
Completed |
|||
Azacitidine versus Decitabine |
II |
MDS |
NCT02269280 |
Recruiting |
|
II |
MDS |
NCT01720225 |
Recruiting |
||
III |
MDS |
NCT01409070 |
Completed |
||
IV |
MDS |
NCT01011283 |
Terminated |
||
SGI-110 |
I |
AML |
NCT02293993 |
Recruiting |
|
I/II |
AML, CMML, MDS |
NCT01261312 |
Ongoing |
||
II |
Idarubicin, Cladribine |
AML |
NCT02096055 |
Recruiting |
|
MDS |
NCT02131597 |
Recruiting |
|||
MDS |
NCT02197676 |
Recruiting |
|||
III |
AML |
NCT02348489 |
Recruiting |
a AHM, advanced hematologic cancers; AML, acute myeloid leukemia; ATRA, all-trans retinoic acid; CCR, Conventional Care Regimen; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; HL, Hodgkin's lymphoma; JMML, juvenile myelomonocytic leukemia; MDS, myelodysplastic sindrome; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; SLL, small lymphocytic lymphoma; VA, valproic acid.
b Identifier of the trial as retrieved April, 2016, from: http://clinicaltrials.gov.
Initially, these drugs were used at relatively high doses, which however turned out to be toxic and did not show a satisfactory antitumor activity. By contrast, lower doses of azacitidine and 5-AZA-CdR resulted in a stronger DNA hypomethylating activity and a better antineoplastic effect [103].
The initial trials that demonstrated the efficacy of azacitidine started in 1984 and were carried out by the Cancer and Leukemia Group B (CALGB). In the first phase II study, named CALGB 8421, 48 patients with MDS were enrolled and treated with monthly azacitidine, 75 mg/m2/day continuous infusion for 7 days. Complete response (CR) was seen in 15% of azacitidine -treated patients, partial remission (PR) was rare, and 21% of patients showed hematological improvement (HI) [104]. Similar response rates were observed in the subsequent CALGB 8921 trial that included 70 patients with MDS, treated with either intravenous or subcutaneous azacitidine (75 mg/m2/d for 7 days every 28 days) [104]. In the last phase III randomized clinical trial CALGB 9221, a total of 191 patients with MDS were randomized to receive azacitidine 75 mg/m2/day subcutaneously for 7 days every 28 days, or supportive care. The results demonstrated a 60% overall response for MDS patients in the azacitidine arm compared to those treated with supportive care only. In particular, in MDS patients treated with low-dose azacitidine, CR, PR and HI were documented in 7%, 16% and 37% of cases, respectively. Furthermore, the median time-to-progression to AML or death was 13 months for patients in the supportive care arm compared with 21 months for patients randomly assigned to azacitidine, with significantly improved quality of life [104]. These results led to azacitidine approval by the FDA for the treatment of MDS. These three CALGB trials included a large number of patients with refractory anemia with excess blasts in transformation that, according to World Health Organization (WHO) criteria, was reclassified as AML with multilineage dysplasia, and with blasts between 20% and 30% [104]. The impact of azacitidine on the survival of MDS patients was further investigated in the international phase III AZA-001 trial. A total of 358 patients with intermediate-2 or high-risk MDS according to the International Prognostic Scoring System (IPSS) were randomized to receive either azacitidine or a conventional care regimen (CCR) that included either low-dose cytarabine, conventional AML induction chemotherapy or supportive care. The results obtained from the phase III randomized AZA-001 trial demonstrated that azacitidine significantly prolonged overall survival (OS) in higher-risk MDS compared with CCR. In fact, patients treated with azacitidine achieved a CR or PR of 29% versus 12%, a two-year OS of 51% versus 26% and a median time to AML transformation or death of 13 months versus 7.6 months when compared with the CCR group. Furthermore, CR was not necessary to prolong OS, since patients with HI had similar two-year survival rates [105]. In March 2009, azacitidine was also approved by the European Medicines Agency for patients who are not eligible for allogeneic hematopoietic stem cell transplantation with the following diagnoses: intermediate- or high-risk MDS according to IPSS, or chronic myelomonocytic leukemia with 10%-29% blasts without myeloproliferative disorder, or patients with AML with 20%- 30% blasts and multilineage dysplasia according to the WHO classification [106]. In a recent large phase III multicenter randomized trial, the AZA-AML-001 study, 488 AML patients aged 65 years or older, with newly-diagnosed or secondary AML with >30% bone marrow blasts and white blood cell counts ≤15 × 109/L were randomized to receive either azacitidine (75 mg/m2/day for seven days subcutaneously of each 28-day cycle) or CCR. Interestingly, in the larger group of older AML patients with >30% bone marrow blasts (n = 480), azacitidine prolonged median OS compared with CCR by 3.8 months (10.4 versus 6.5 months) [107].
5-AZA-CdR has been first explored in several phase I/II studies, which showed safety and efficacy and reported encouraging results in AML and MDS patients. These initial data, together with the reported activity of low concentrations of 5-AZA-CdR to induce cellular differentiation in vitro, provided the rationale to investigate the activity and toxicity of lower doses of 5-AZA-CdR in further studies (for review see [108, 109, 110). These results led to the first phase III randomized trial of 5-AZA-CdR compared with supportive care in 170 MDS patients, which formed the basis for the FDA approval of decitabine. Patients were randomly assigned to receive either supportive care or decitabine at a dose of 15 mg/m2 as a 3-hour infusion every 8 hours for 3 days, repeated every 6 weeks, plus supportive care. 5-AZA-CdR was clinically effective in the treatment of patients with MDS, providing durable responses and improving time to AML transformation or to death, with an observed response rate of 17% {Kantarjian, 2006 #1191]. In a subsequent study, 22 MDS patients who had previously responded to low-dose 5-AZA-CdR were re-treated at the time of disease relapse at a median of 11 months after the last course of initial treatment [111]. Results showed that 45% patients were still responsive, indicating persistent sensitivity to the drug. However, the duration of the second response was inferior to that of the first remission and upfront resistance to the second treatment was also noted, suggesting that a longer period of initial treatment might result in an increased clinical benefit [111]. To address this issue, other 5-AZA-CdR schedules have been evaluated and further improvement was observed with continued treatment. Kantarjian et al. carried out a randomized phase III study, including a 5-day schedule at 20 mg/m2 intravenously (IV), a 5-day schedule at 20mg/m2 subcutaneously, and a 10-day schedule at 10 mg/m2 IV, in 95 patients with MDS and CMML. Each course of decitabine was delivered every 4 weeks and response or failure of therapy were evaluated after at least three courses. The 5-day IV schedule was considered the optimal schedule, since it gave the highest CR rate [112]. Similar results were observed in a phase II study in patients with AML treated with decitabine 20 mg/m2 IV for 5 consecutive days of a 4-week cycle [113]. Despite the success of azacitidine and 5-AZA-CdR in the treatment of hematologic malignancies, the in vivo mechanisms of these DNMTi still need to be elucidated to guide further trials. The metabolic instability of azacitidine and 5-AZA-CdR has prompted the development of modified analogs, including SGI-110, a dinucleotide that includes a deoxyguanosine and is largely resistant to cytidine deaminase activity [114]. SGI-110 has shown a good tolerability in vivo [115] and antitumor activities in xenograft models and in primates [114, 116, 117]. SGI-110 is currently being studied in a randomized phase I-II-III clinical trials focused on treatment of patients with AML and MDS with promising results [118] (Table 1). Several phase I studies with 5-AZA-CdR have also been conducted in NHL patients, but the response to therapy was moderate [119-121]. Other clinical trials are currently testing the ability of DHA to potentiate the activity of conventional chemotherapeutic agents in hematological malignancies.
IDH inhibitors
Early clinical data obtained with IDH inhibitors suggested their activity in patients with advanced hematologic malignancies, including relapsed/refractory AML and MDS. AG-6780 is a small molecular inhibitor of mutant IDH2 Treatment with AGI-6780 was able to stimulate the expression of maturation markers in leukemic cells in vitro, indicating the induction of differentiation [122]. AG-221, another inhibitor of mutant IDH2, has demonstrated selective and sensitive effects on AML cells harboring IDH2 mutations both in vitro and in vivo. AG-221 has been recently introduced into phase I/II clinical trials for patients with AML and angioimmunoblastic T-cell lymphoma with encouraging preliminary results [123].
HDAC inhibitors
Given that increased HDAC expression and activity are found is many hematological malignancies, HDAC inhibitors (HDACi) have been extensively used for the treatment of B- and T-cell malignancies (Table 2). Based on their chemical structures, HDACi have been divided into several classes: short-chain fatty acids, hydroxamic acids and hydroxamic acid-based hybrid polar compounds, cyclic tetrapeptides, benzamides and miscellaneous compounds. The antineoplastic activity of these drugs is related to altered gene expression and to changes in non-histone proteins in virtually all most cancer-related pathways. Evidence accumulated so far indicates that the therapeutic potential of HDACi probably stems from their ability to induce selective cell cycle arrest, differentiation and/or apoptosis in different cell types by modulating the expression of target genes [79]. To date, several HDACi have been approved for cancer therapy by the FDA.
Table 2: Summary of clinical trials examining Histone Deacetylase, Histone Methyltransferase, and Bromodomain inhibitors in hematological malignancies
HDAC inhibitor |
Phase |
Combination |
Hematological disease |
Identifier |
State |
4SC-202 |
I |
AHM |
NCT01344707 |
Completed |
|
Abexinostat |
I |
HL, NHL, MM |
NCT01149668 |
Completed |
|
I/II |
HL, FL, MCL |
NCT00724984 |
Completed |
||
Belinostat |
I |
CHOP |
PTCL |
NCT01839097 |
Ongoing |
I |
HL, B-cell, T-cell or NK-NHL |
NCT00413075 |
Completed |
||
II |
PTCL |
NCT00865969 |
Completed |
||
II |
MDS |
NCT00357162 |
Completed |
||
II |
MM |
NCT00131261 |
Completed |
||
II |
AML |
NCT00357032 |
Completed |
||
II |
B-cell-NHL |
NCT00303953 |
Completed |
||
II |
CTCL, NHL, PTCL |
NCT00274651 |
Terminated |
||
CI-994 |
II |
MM |
NCT00005624 |
Completed |
|
CUDC-907 |
I |
HL, MM, NHL |
NCT01742988 |
Recruiting |
|
Entinostat |
I |
HL, NHL |
NCT00020579 |
Completed |
|
I |
ALL, AML, CML, MDS, MM |
NCT00015925 |
Completed |
||
II |
HL |
NCT00866333 |
Ongoing |
||
Givinostat |
I/II |
HL |
NCT00496431 |
Terminated |
|
II |
CLL |
NCT00792831 |
Terminated |
||
ITF2357 |
II |
MM |
NCT00792506 |
Terminated |
|
Mocetinostat |
I/II |
DLBCL, FL |
NCT02282358 |
Recruiting |
|
II |
HL |
NCT00358982 |
Terminated |
||
Panobinostat |
I |
Everolimus |
HL, MM, NHL |
NCT00962507 |
Completed |
I |
CTCL |
NCT00412997 |
Completed |
||
I |
HL, NHL |
NCT01032148 |
Recruiting |
||
I |
AML |
NCT01242774 |
Completed |
||
I |
HL |
NCT00742027 |
Completed |
||
I |
MM |
NCT00532389 |
Completed |
||
I/II |
Everolimus |
HL, MM, NHL |
NCT00918333 |
Ongoing |
|
I/II |
AHM |
NCT00621244 |
Completed |
||
I/II |
AML, MDS |
NCT01451268 |
Recruiting |
||
II |
Rituximab |
DLBCL |
NCT01282476 |
Ongoing |
|
II |
Rituximab |
DLBCL |
NCT01238692 |
Ongoing |
|
II |
NHL |
NCT01261247 |
Ongoing |
||
II |
NHL |
NCT01090973 |
Terminated |
||
II |
AML |
NCT00880269 |
Completed |
||
II |
NHL |
NCT01090973 |
Terminated |
||
II |
CTCL, Adult T-cell Leukemia/Lymphoma |
NCT00699296 |
Terminated |
||
II |
ALL, AML |
NCT00723203 |
Terminated |
||
II |
MDS |
NCT00939159 |
Terminated |
||
II |
MDS |
NCT00594230 |
Terminated |
||
II |
MM |
NCT00445068 |
Terminated |
||
II/III |
CTCL |
NCT00490776 |
Completed |
||
II/III |
CML |
NCT00451035 |
Completed |
||
II/III |
CML |
NCT00449761 |
Completed |
||
II/III |
CTCL |
NCT00425555 |
Completed |
||
Pivanex |
II |
CLL |
NCT00083473 |
Terminated |
|
Quisinostat |
I |
MDS |
NCT00676728 |
Terminated |
|
II |
CTCL |
NCT01486277 |
Ongoing |
||
Resminostat |
II |
HL |
NCT01037478 |
Completed |
|
Rocilinostat |
I/II |
HL, NL |
NCT02091063 |
Recruiting |
|
I/II |
MM |
NCT01323751 |
Ongoing |
||
Romidepsin |
I/II |
Azacitidine |
HL, NHL |
NCT01998035 |
Recruiting |
II |
CTCL, PTCL |
NCT00007345 |
Ongoing |
||
II |
NHL |
NCT00077194 |
Completed |
||
II |
NHL |
NCT00383565 |
Terminated |
||
SHAPE |
II |
CTCL |
NCT02213861 |
Recruiting |
|
SHP-141 |
I |
CTCL |
NCT01433731 |
Completed |
|
Tefinostat |
I |
AML, CLL, CML CMML, HL, MDS, MM, NHL |
NCT00820508 |
Completed |
|
VPA |
II |
CLL |
NCT00524667 |
Terminated |
|
Valproate |
I/II |
Rituximab, CHOP |
DLBCL |
NCT01622439 |
Recruiting |
I/II |
MDS |
NCT00776503 |
Completed |
||
Vorinostat |
I |
B-cell lymphomas excluding CLL |
NCT01276717 |
Completed |
|
I |
CTCL |
NCT00771472 |
Completed |
||
I |
HL, NHL |
NCT00127140 |
Completed |
||
I |
AML, CLL, CML CMML, HL, MDS, MM, NHL |
NCT00005634 |
Completed |
||
I |
Lenalidomide |
HL, NHL |
NCT01116154 |
Terminated |
|
I/II |
Rituximab, CHOP |
DLBCL |
NCT00972478 |
Ongoing |
|
I/II |
Cyclophosphamide, Etoposide, Prednisone, Rituximab |
DLBCL |
NCT00667615 |
Ongoing |
|
I/II |
Rituximab, Ifosfamide, Carboplatin, Etoposide |
MCL, NHL |
NCT00601718 |
Completed |
|
I/II |
CHOP |
NHL |
NCT00787527 |
Completed |
|
I/II |
MM |
NCT00857324 |
Terminated |
||
II |
Bortezomib |
DLBCL, MCL |
NCT00703664 |
Ongoing |
|
II |
B-NHL, FL, MCL |
NCT00875056 |
Ongoing |
||
II |
NHL |
NCT00253630 |
Ongoing |
||
II |
NHL |
NCT00077194 |
Completed |
||
II |
AML |
NCT00305773 |
Completed |
||
II |
HL |
NCT00132028 |
Completed |
||
II |
CTCL |
NCT00958074 |
Terminated |
||
II |
MDS |
NCT00486720 |
Terminated |
||
HMT inhibitor |
Phase |
Hematological disease |
Identifier |
State |
|
EPZ-5676 |
I |
ALL, AML |
NCT02141828 |
Ongoing |
|
I |
ALL, AML, MDS |
NCT01684150 |
Ongoing |
||
EPZ-6438 |
I/II |
B-cell lymphomas, DLBCL, FL |
NCT01897571 |
||
BET inhibitor |
Phase |
Hematological disease |
Identifier |
State |
|
FT-1101 |
I |
AML, MDS |
NCT02543879 |
Recruiting |
|
CPI-0610 |
I |
HL, NHL |
NCT01949883 |
Recruiting |
|
I |
MM |
NCT02157636 |
Recruiting |
||
I |
ALL, AML, CML, MDS |
NCT02158858 |
Recruiting |
||
GSK525762 |
I/II |
AML, NHL, MM |
NCT01943851 |
Recruiting |
|
OTX015 |
I |
AML, ALL, DLBCL, MM |
NCT01713582 |
Ongoing |
a AHM, advanced hematologic malignancies; AML, acute myeloid leukemia; BET, bromodomain and extra-terminal motif; CHOP, Cyclophosphamide/Vincristine/Doxorubicin/Prednisone; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; CTCL, cutaneous T-cell lymphoma; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; HDAC, histone deacetylase; HL, Hodgkin's lymphoma; HMT, histone methyltransferase; MCL, mantle cell lymphoma; MDS, myelodysplastic syndrome; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; PTCL, Peripheral T-Cell Lymphoma; VA, valproic acid.
b Identifier of the trial as retrieved April, 2016, from http://clinicaltrials.gov.
Short-chain fatty acids
These compounds represent a class of HDACi with simple structures that have shown clinical potential in various studies. Valproic acid (VPA) and phenylbutyrate are two well characterized compounds that belong to this class of compounds. VPA has been increasingly studied in clinical trials as a single agent or in combination therapies. VPA exerts a wide range of effects on AML and results from previous studies have clearly demonstrated that VPA has antiproliferative and pro-apoptotic effects on AML cells [124]. In a phase II study of AML and MDS, 66 patients were initially treated with VPA alone with the later addition of all-trans-retinoic acid (ATRA) in non-responsive or relapsed patients. HI was observed in 24% of patients, with a median response duration of 4 months [125]. In a phase I/II study, 44 AML and MDS patients were treated with escalating doses of VPA and concomitantly with a fixed dose of 5-AZA-CdR. Global DNA hypomethylation and histone H3 and H4 acetylation were associated with p15 reactivation in PBMC from patients. However, no correlation was found between these epigenetic modifications and the clinical response. Furthermore, the observed response rate (22%) was much lower than that observed in trials in which 5-AZA-CdR was used alone, thus suggesting that VPA did not offer a significant improvement to the response [126]. VPA has been used in clinical trials for AML and MDS, also in combination with ATRA and azacitidine. In a trial conducted by Soriano et al., 49 AML and 5 MDS patients were treated with a high-dose intermittent schedule of VPA (50 mg/kg for 7 days) combined with the approved azacitidine dosage and ATRA given at 45 mg/m2 for 5 days. The combination was safe and clinically active, with an overall response rate of 42%, and 12 (22%) CR. Furthermore, global DNA hypomethylation and histone acetylation were observed but they did not correlate with the clinical response, thus suggesting that responses to HDACi are likely related to non-histone acetylation or to other mechanisms [127]. A further study combining azacitidine (75 mg/m2) and VPA (35-50 mg/kg) for 7 days followed by ATRA (45 mg/m2) for 21 days was conducted in patients with high-risk AML or MDS. Among the 65 patients enrolled in this study, 14 showed a PR and 3 a CR. Interestingly, promoter demethylation of four genes (FZD9, ALOX12, HPN, and CALCA) was associated with the clinical response [128]. A shortened azacitidine schedule (75 mg/m2 for 5 days) plus VPA, ATRA, and theophylline was evaluated in patients with AML or MDS. In 15 out of 79 patients achieving CR, leukemic stem cells were substantially reduced, but never eradicated, since expansion of this population took place before morphological relapse [129], thus suggesting that treatment interruption might be associated with rapid relapse.
Hydroxamic acids
Hydroxamic acids include the majority of HDACi currently employed in clinical trials for the treatment of hematological diseases. Among the hydroxamic acids, vorinostat (SAHA, Zolina) was approved by the FDA in 2006 for the treatment of patients with cutaneous T-cell lymphoma (CTCL), a rare type of NHL of the skin. In pivotal phase II clinical trial, the daily treatment of refractory CTCL patients with 400 mg of vorinostat showed a median duration of response of approximately 185 days with a good tolerability and safety profile [130]. Vorinostat treatment was also safe and effective in another phase II clinical trial that involved patients with relapsed/refractory NHL and mantle cell lymphoma [131]. At the molecular level, the antiproliferative effect of vorinostat leads to accumulation of acetylated histones, p21, BAX, STAT6, and caspases, resulting in cell cycle arrest, growth inhibition, apoptosis, and differentiation of cells from AML, MDS, and CTCL patients [132, 133]. Preclinical studies combining rituximab and vorinostat reported increased rituximab activity in B-cell NHL by preventing NF-kB nuclear translocation and promoting its degradation [134]. This combination was further evaluated in a phase II clinical trial in 28 patients with indolent B-cell NHL. The regimen was well-tolerated and appeared promising, with a median progression-free survival (PFS) of 29.2 months for all patients, 18.8 months for relapsed/refractory patients; PFS was not reached in untreated patients [135]. Vorinostat is currently being investigated alone or in combination therapies in several clinical trials for different hematological malignancies (for review, see [136]).
Besides vorinostat, panobinostat (LBH589), belinostat (PDX-101, and pracinostat (SB939), represent a second generation of hydroxamate-based compounds. Panobinostat was recently approved by the FDA for use in combination with bortezomib and dexamethasone in patients with MM. The approval was based on findings in patients with relapsed or relapsed and refractory MM who participated in a clinical trial called PANORAMA1 [137] Panobinostat potently induced cell cycle arrest, apoptosis, and H3K9 and H4K8 hyperacetylation in ALL [138]. Panobinostat was shown to improve the antileukemic effect of fludarabine through a predominantly apoptotic mechanism. The synergism is striking in vitro in cell lines, in patient cells, but also in an in vivo xenograft model of AML [139]. Furthermore, panobinostat sensitizes leukemic blasts to treatment with cytarabine and daunorubicin by suppressing the expression of BRCA1, CHK1, and RAD51 through transcriptional mechanisms [140]. A phase I/II study combining panobinostat plus decitabine in ederly patients with high risk MDS or AML is currently evaluating the efficacy and safety of this combination [118, 141]. Panobinostat is also under investigation in a phase I/II study in combination with azacitidine in adult patients with MDS, CMML, or AML [118]. Belinostat was approved on 2014 for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL) upon results from the phase II BELIEF study [142]. The anticancer effect of belinostat is mediated through the acetylation of H3 and H4, which was seen both in vivo and in vitro after belinostat exposure. Cancer cell growth inhibition and apoptosis were associated with these increased levels of acetylation [143]. Belinostat is currently being evaluated in phase I/II trials in CTCL and PTCL (for review see [144]), whereas it has shown only minimal activity in AML [145] and no effect in MDS [146]. In DLBCL and PTCL models, belinostat has also been investigated in combination with DNMTi, and synergistic action was observed [147].
Pracinostat gained Orphan Drug status by the FDA in 2014 for future development in AML. In a phase I study of older AML patients, pracinostat induced prolonged CRR of 14% lasting for 206 and 362 days. A phase II trial of pracinostat in combination with azacitidine in patients with high-risk MDS yielded an 89% objective response rate, including 78% CR or CR with insufficient blood count recovery and 56% complete cytogenetic responses. Based on these encouraging results, several studies are currently recruiting patients with MDS or AML [118, 148].
Benzamides
Benzamides are synthetic compounds with efficient HDAC inhibitory activity that is mediated by the targeting of the Zn2+ ion in the catalytic pocket of the enzyme. [149]. Entinostat (MS-275) and mocetinostat (MGCD0103) are under clinical evaluation as single agents and in combination with other drugs. Entinostat has been also combined with azacitidine in patients with AML and MDS, but the overlapping schedule of this combination was not superior to azacitidine monotherapy and was associated with pharmacodynamic antagonism [150]. In preclinical studies in HL, entinostat increased H3 acetylation, up-regulated p21 expression, and promoted the intrinsic apoptosis pathway by downregulating Bcl-2 and Bcl-xL proteins [151]. In in vivo experiments, entinostat was also found to enhance the antitumor activity of rituximab-sensitive and -resistant B-cell lymphoma cell lines [152].
Cyclic peptides
Several cyclic peptides isolated from microorganisms have been described to possess HDAC inhibitory activity. Romidepsin (Istodax, FK228, FR901228, depsipeptide), a natural product obtained from the bacterium Chromobacterium violaceum, was approved by the FDA in 2009 for the treatment of the refractory form of CTCL. The FDA approval of romidepsin for the treatment of CTCL was based on two large phase II studies: a multi-institutional study and an international study including 71 and 96 patients, respectively. The treatment schedule was identical and produced overall response rates of 34-35%, with a CR in about 6% of patients in both studies. The median duration of response was 13.7 months [153]. In 2011, romidepsin was also approved for PTCL in patients who had received at least one prior therapy. In the pivotal study, including 130 patients with relapsed or refractory PTCL, objective responses were seen in 25% and CR in 15% [154]. The median duration of response was 28 months with a median follow-up of 22.3 months [155]. In patients with CLL, AML and MDS, romidepsin did not show significant clinical efficacy, but exhibited some biological activity as indicated by an increase in p21 protein expression concurrent with H4 acetylation of the p21 promoter gene [156, 157]. Recently, it has been demonstrated that romidepsin also has DNA hypomethylating activity, which might inhibit the Wnt/β-catenin pathway in T-lymphoblastic cells [158]. Romidepsin is currently being evaluated in several studies, either as a single agent or in combination with other drugs for treating mainly T-cell lymphoma.
HAT inhibitors
Unlike HDACs, little is known about inhibitors of HATs (HATi) in B- or T-cell cancers, and no HATi are currently approved by the FDA. HATi may have therapeutic potential and several natural HATi are under evaluation for their anti-cancer properties. Among these natural compounds, curcumin has gained increased attention as a potential anti-cancer drug and has being investigated in a variety of tumors, including MM [159]. Curcumin has been shown to possesses intrinsic HATi activity specific for p300/CBP both in vitro and in vivo and induce cell proliferation arrest and apoptosis [160]. However, curcumin has many protein targets, and thus it is not clear as to whether its effects are only due to its anti-HAT activity [161]. Anacardic acid, a chemical compounds found in the shell of the cashew nut, is a potent inhibitor of several HAT, including p300, PCAF, and TIP60. Anacardic acid was demonstrated to inhibit the growth of Jurkat T-cell leukemia cells [162], which express two TIP60 variants, including one with a deleted HAT domain [163].
HMT inhibitors
So far, only a few HMT inhibitors (HMTi) are known, and most of them were discovered through random screening approaches. Deazaneplanocin A (DZNep) is an HMTi that has a wide range of anticancer effects in several human cancers. In particular, DZNep has been found to deplete EZH2 levels and to inhibit H3K27 trimethylation in AML cell lines in a dose-dependent manner. DZNep treatment increases both mRNA and protein levels of p16, p21, and p27 in AML cells, causing cell cycle arrest and apoptosis. Co-treatment with panobinostat enhances the antileukemic effects of DZNep and was found to synergistically improve the survival of mice implanted with AML cells [164]. Two other EZH2 inhibitors, GSK126 and EPZ005687, have shown preferential effectiveness in suppressing the growth of lymphoma-associated mutants of EZH2 in comparison to those with wild-type EZH2 [165, 166]. In a study by McCabe et al, GSK126 inhibited the proliferation of Y641 EZH2 DLBCL cell lines and induced a 50% loss of H3K27 trimethylation. Besides cell-based studies, treatment of xenografted DLBCL models resulted in either tumor regression or tumor inhibition, depending on the dosing regimen, and GSK126 was well tolerated [165]. Similarly, EPZ0056687 has been shown to induce apoptotic cell killing in heterozygous mutant EZH2 Y641 or alanine 677 lymphoma cells, with minimal effects on wild-type cell proliferation [166]. EPZ004777 was the first specific DOT1L small molecular inhibitor to be been extensively characterized. Daigle et al. reported decreased H3K79 methylation levels and selective inhibition of leukemogenic genes expression in MLL cells exposed to EPZ004777. Furthermore, administration of EPZ004777 resulted in selective apoptosis of cell lines harboring MLL gene translocation, with minimum effect on non-MLL-rearranged cells. Finally, in vivo EPZ004777 treatment increased overall survival in an MLL xenograft model [167]. EPZ-5676 is another potent and selective DOT1L inhibitor which selectively kills cells containing the MLL chromosomal translocation, whereas it shows little effect on leukemia cells that lack this translocation. In a rat xenograft model of MLL-rearranged leukemia, EPZ- 5676 was able to induce significant cancer growth inhibition. Furthermore, complete regression was achieved following 21 days of continuous intravenous administration of EPZ-5676 and tumor regrowth was not observed until the end of the experiment [168]. EPZ-5676 has now advanced to phase I clinical trials in patients with advanced hematological malignancies, including AML with MLL fusions (Table 2).
BET inhibitors
In 2010, two independent research groups demonstrated the excellent activity of two BET proteins small-molecule inhibitors, known as I-BET762 (GSK 525762A) and JQ1 [169, 170]. During a screening that evaluated the ability of synthetic compounds to bind selectively to individual proteins in cell lysates, I-BET762 showed the highest affinity interaction with the acetyl-lysine binding site of BRD4-BD1. In this study, I-BET762 significantly improved the survival of C57BL/6 mice with severe sepsis by disrupting chromatin complexes responsible for the expression of crucial inflammatory genes [169]. The inhibitor JQ1 was initially shown to have efficacy in the context of nuclear protein in testis (NUT) midline carcinoma (NMC). In the majority of NMC the two N-terminal BET bromodomains of BRD4 are fused with NUT via the t(15;19) translocation to create an oncogenic fusion gene whose product is driven by the BRD4 promoter. Treatment of NMC cell lines and xenograft models with JQ1 displaced the BRD4 fusion protein from nuclear chromatin and induced squamous differentiation, thus exhibiting specific anti-proliferative effects [170]. The results of I-BET762 and JQ1 treatments have encouraged the development of other BET inhibitors with clear antiproliferative and pro-apoptotic effects in several hematological malignancies, including MM [171], NHL [172], AML [95, 173] and ALL [174]. Based on the observed down-regulation of a common transcriptional program that includes critical oncogenic targets such as BCL2 and c-MYC, several phase I clinical trials have been initiated to explore the efficacy of BET inhibitors in AML and other hematological malignancies (Table 2). The first published evidence of the clinical activity of BET inhibitors refers to a phase I clinical trial with OTX015, in which promising antitumor activity was seen in both acute leukemia and other hematological malignancies (Patrice Herait, AACR Annual Meeting, San Diego, LA, USA; Oral communication, April, 2014).
CONCLUSIONs
Somatic alterations in genes involved in DNA methylation and histone modifications have emerged central events in the development and progression of hematological malignancies. Loss or gain of function mutations affecting catalytic domains deregulate the activity of epigenetic enzymes and may promote leukemogenesis by altering the normal self-renewal and differentiation of HSC. Furthermore, fusion proteins generated by translocation often mediate their oncogenic potential by directly or indirectly interfering with epigenetic modifying activities. However, the epigenetic mechanisms exploited by leukemic fusion proteins to drive the induction and maintenance of the leukemic state are still far from being elucidated. So far, DHA and HDACi have been widely used to evaluate the potential of epigenetic modulation to benefit patient outcome. Nevertheless, the currently available epigenetic drugs are non-specific, so new therapeutic molecules with well-defined targeting of the epigenetic machinery should be developed. Rational combination of available epigenetic drugs with targeted therapies also warrants investigation in the clinical setting. Further characterization of the genetics of hematological malignancies, together with a better understanding of the role of epigenetic alterations in leukemic transformation will probably provide amenable targets for next-generation epigenetic drugs.
ACKNOWLEDGMENTS
The authors thank Mrs. Anna Vallerugo, M.A., for her writing assistance. This study was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (contract 14287 to RD) and from the Centro di Riferimento Oncologico I.R.C.C.S. (Intramural grant to EF and LS).
CONFLICT OF INTEREST
No conflict of interest to be disclosed.
References
1. Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D and Ebert BL. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011; 364:2496-2506.
2. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, Shiraishi Y, Shiozawa Y, Chiba K, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014; 28:241-247.
3. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009; 361:1058-1066.
4. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, et al. Prognostic Relevance of Integrated Genetic Profiling in Acute Myeloid Leukemia. The New England journal of medicine. 2012; 366:1079-1089.
5. Illingworth RS and Bird AP. CpG islands--’a rough guide’. FEBS Lett. 2009; 583:1713-1720.
6. Smith ZD and Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013; 14:204-220.
7. Kulis M and Esteller M. DNA methylation and cancer. Adv Genet. 2010; 70:27-56.
8. Dahl C, Gronbaek K and Guldberg P. Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin Chim Acta. 2011; 412:831-836.
9. Goodell MA and Godley LA. Perspectives and future directions for epigenetics in hematology. Blood. 2013; 121:5131-5137.
10. Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, Nerlov C, Leutz A, ndrade-Navarro MA, Jacobsen SE and Rosenbauer F. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009; 41:1207-1215.
11. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, Liang S, Lu Y, Darlington GJ, Meissner A, Issa JP, Godley LA, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012; 44:23-31.
12. Ostronoff F, Othus M, Ho PA, Kutny M, Geraghty DE, Petersdorf SH, Godwin JE, Willman CL, Radich JP, Appelbaum FR, Stirewalt DL and Meshinchi S. Mutations in the DNMT3A exon 23 independently predict poor outcome in older patients with acute myeloid leukemia: a SWOG report. Leukemia. 2013; 27:238-241.
13. Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, Rockova V, Sanders M, Abbas S, Figueroa ME, Zeilemaker A, Melnick A, Lowenberg B, Valk PJ and Delwel R. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012; 119:5824-5831.
14. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, et al. DNMT3A Mutations in Acute Myeloid Leukemia. The New England journal of medicine. 2010; 363:2424-2433.
15. K.Thirukonda V, Raghupathy R and Parekh S. Translational Medicine. 2012.
16. Leone G, Teofili L, Voso MT and Lubbert M. DNA methylation and demethylating drugs in myelodysplastic syndromes and secondary leukemias. Haematologica. 2002; 87:1324-1341.
17. Rosu-Myles M and Wolff L. p15Ink4b: dual function in myelopoiesis and inactivation in myeloid disease. Blood Cells Mol Dis. 2008; 40:406-409.
18. Khan H, Vale C, Bhagat T and Verma A. Role of DNA methylation in the pathogenesis and treatment of myelodysplastic syndromes. Semin Hematol. 2013; 50:16-37.
19. Hopfer O, Komor M, Koehler IS, Freitag C, Schulze M, Hoelzer D, Thiel E and Hofmann WK. Aberrant promotor methylation in MDS hematopoietic cells during in vitro lineage specific differentiation is differently associated with DNMT isoforms. Leuk Res. 2009; 33:434-442.
20. Schulz WA, Elo JP, Florl AR, Pennanen S, Santourlidis S, Engers R, Buchardt M, Seifert HH and Visakorpi T. Genomewide DNA hypomethylation is associated with alterations on chromosome 8 in prostate carcinoma. Genes Chromosomes Cancer. 2002; 35:58-65.
21. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002; 21:5400-5413.
22. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Cervantes F, Sanchez J, Garate L, Barrios M, Castillejo JA, Navarro G, Colomer D, Prosper F, Heiniger A and Torres A. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene. 2005; 24:7213-7223.
23. Delhommeau F, Dupont S, Della V, V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, Lecluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009; 360:2289-2301.
24. Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gäken J, Lea NC, Przychodzen B, Mian SA, Nasser EE, Shooter C, Westwood NB, Strupp C, Gattermann N, Maciejewski JP, Germing U and Mufti GJ. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood. 2010; 116:3923-3932.
25. Butler JS and Dent SY. The role of chromatin modifiers in normal and malignant hematopoiesis. Blood. 2013; 121:3076-3084.
26. Fong CY, Morison J and Dawson MA. Epigenetics in the hematologic malignancies. Haematologica. 2014; 99:1772-1783.
27. Rothbart SB and Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014; 1839:627-643.
28. Marzluff WF and Duronio RJ. Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol. 2002; 14:692-699.
29. Talbert PB and Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010; 11:264-275.
30. Marzluff WF, Wagner EJ and Duronio RJ. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet. 2008; 9:843-854.
31. Weber CM, Ramachandran S and Henikoff S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol Cell. 2014; 53:819-830.
32. Conerly ML, Teves SS, Diolaiti D, Ulrich M, Eisenman RN and Henikoff S. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res. 2010; 20:1383-1390.
33. Je EM, Yoo NJ, Kim YJ, Kim MS and Lee SH. Somatic mutation of H3F3A, a chromatin remodeling gene, is rare in acute leukemias and non-Hodgkin lymphoma. European Journal of Haematology. 2013; 90:169-170.
34. Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF and Bernstein E. Histone variants: emerging players in cancer biology. Cell Mol Life Sci. 2014; 71:379-404.
35. Novik KL, Spinelli JJ, Macarthur AC, Shumansky K, Sipahimalani P, Leach S, Lai A, Connors JM, Gascoyne RD, Gallagher RP and Brooks-Wilson AR. Genetic variation in H2AFX contributes to risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2007; 16:1098-1106.
36. Morgan MA and Shilatifard A. Chromatin signatures of cancer. Genes Dev. 2015; 29:238-249.
37. Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, Scott DW, Ding J, Roth A, Chiu R, Corbett RD, Chan FC, Mendez-Lago M, Trinh DL, Bolger-Munro M, Taylor G, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013; 122:1256-1265.
38. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, Fox H, Jacobi K, Saiya-Cork K, Bixby D, Lebovic D, Roulston D, Shedden K, Sabel M, Marentette L, Cimmino V, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014; 123:1487-1498.
39. Greer EL and Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012; 13:343-357.
40. Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE and Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002; 111:197-208.
41. Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, Martinez-Garcia E, Zhang H, Zheng Y, Verma SK, McCabe MT, Ott HM, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013; 23:677-692.
42. Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM and Elemento O. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010; 116:5247-5255.
43. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, et al. Somatic mutation of EZH2 (Y641) in Follicular and Diffuse Large B-cell Lymphomas of Germinal Center Origin. Nature genetics. 2010; 42:181-185.
44. Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, Marquez VE, Marra MA, Gascoyne RD, Humphries RK, Arrowsmith CH, Morin GB, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011; 117:2451-2459.
45. Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM and Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010; 107:20980-20985.
46. Popovic R, Shah MY and Licht JD. Epigenetic therapy of hematological malignancies: where are we now? Therapeutic Advances in Hematology. 2013; 4:81-91.
47. Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, Beran M, Gilliland DG, Levine RL and Tefferi A. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011; 25:1200-1202.
48. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der HA, Scheele TN, Vandenberghe P, de WT, van der Reijden BA and Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010; 42:665-667.
49. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010; 42:722-726.
50. Jaju RJ, Fidler C, Haas OA, Strickson AJ, Watkins F, Clark K, Cross NC, Cheng JF, Aplan PD, Kearney L, Boultwood J and Wainscoat JS. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood. 2001; 98:1264-1267.
51. Wang GG, Cai L, Pasillas MP and Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol. 2007; 9:804-812.
52. Keats JJ, Reiman T, Belch AR and Pilarski LM. Ten years and counting: so what do we know about t(4;14)(p16;q32) multiple myeloma. Leuk Lymphoma. 2006; 47:2289-2300.
53. Huang Z, Wu H, Chuai S, Xu F, Yan F, Englund N, Wang Z, Zhang H, Fang M, Wang Y, Gu J, Zhang M, Yang T, Zhao K, Yu Y, Dai J, et al. NSD2 is recruited through its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res. 2013; 73:6277-6288.
54. Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E and Young RA. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A. 2005; 102:8603-8608.
55. Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009; 94:984-993.
56. Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, Ben Abdelali R, Macintyre E, De Braekeleer E, De Braekeleer M, Delabesse E, de Oliveira MP, Cave H, Clappier E, van Dongen JJM, Balgobind BV, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009; 23:1490-1499.
57. Dou Y and Hess JL. Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int J Hematol. 2008; 87:10-18.
58. Krivtsov AV and Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007; 7:823-833.
59. Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, Dias S, Chang J, Olhava EJ, Daigle SR, Richon VM, Pollock RM and Armstrong SA. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013; 121:2533-2541.
60. Cheung N, Chan LC, Thompson A, Cleary ML and So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007; 9:1208-1215.
61. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012; 109:3879-3884.
62. Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, Rossi D, Chadburn A, Murty VV, Mullighan CG, Gaidano G, Rabadan R, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011; 471:189-195.
63. Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ, Irish J, Stehr H, Vicente-Duenas C, Romero-Camarero I, Sanchez-Garcia I, Plevritis SK, Arber DA, Batzoglou S, Levy R and Alizadeh AA. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A. 2015; 112:E1116-E1125.
64. Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, Lugar PL, Rizzieri DA, Lagoo AS, Bernal-Mizrachi L, Mann KP, Flowers C, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110:1398-1403.
65. Lakshmikuttyamma A, Takahashi N, Pastural E, Torlakovic E, Amin HM, Garcia-Manero G, Voralia M, Czader M, Decoteau JF and Geyer CR. RIZ1 is potential CML tumor suppressor that is down-regulated during disease progression. J Hematol Oncol. 2009; 2:28.
66. Steele-Perkins G, Fang W, Yang XH, Van GM, Carling T, Gu J, Buyse IM, Fletcher JA, Liu J, Bronson R, Chadwick RB, de la CA, Zhang X, Speleman F and Huang S. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev. 2001; 15:2250-2262.
67. Fears S, Mathieu C, Zeleznik-Le N, Huang S, Rowley JD and Nucifora G. Intergenic splicing of MDS1 and EVI1 occurs in normal tissues as well as in myeloid leukemia and produces a new member of the PR domain family. Proc Natl Acad Sci U S A. 1996; 93:1642-1647.
68. Liu L, Shao G, Steele-Perkins G and Huang S. The retinoblastoma interacting zinc finger gene RIZ produces a PR domain-lacking product through an internal promoter. J Biol Chem. 1997; 272:2984-2991.
69. Brooks DJ, Woodward S, Thompson FH, Dos SB, Russell M, Yang JM, Guan XY, Trent J, Alberts DS and Taetle R. Expression of the zinc finger gene EVI-1 in ovarian and other cancers. Br J Cancer. 1996; 74:1518-1525.
70. Graff J and Tsai LH. Histone acetylation: molecular mnemonics on the chromatin. Nat Rev Neurosci. 2013; 14:97-111.
71. Perez-Campo FM, Borrow J, Kouskoff V and Lacaud G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood. 2009; 113:4866-4874.
72. Collins HM, Kindle KB, Matsuda S, Ryan C, Troke PJ, Kalkhoven E and Heery DM. MOZ-TIF2 alters cofactor recruitment and histone modification at the RARbeta2 promoter: differential effects of MOZ fusion proteins on CBP- and MOZ-dependent activators. J Biol Chem. 2006; 281:17124-17133.
73. Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, Cross NC, Glass CK, Cleary ML and Gilliland DG. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell. 2003; 3:259-271.
74. Kindle KB, Troke PJ, Collins HM, Matsuda S, Bossi D, Bellodi C, Kalkhoven E, Salomoni P, Pelicci PG, Minucci S and Heery DM. MOZ-TIF2 inhibits transcription by nuclear receptors and p53 by impairment of CBP function. Mol Cell Biol. 2005; 25:988-1002.
75. Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT and Livingston DM. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci U S A. 2002; 99:14789-14794.
76. Huang W-C, Ju T-K, Hung M-C and Chen C-C. Phosphorylation of CBP by IKK Promotes Cell Growth by Switching the Binding Preference of CBP from p53 to NF-B. Molecular Cell. 2007; 26:75-87.
77. Chan EM, Chan RJ, Comer EM, Goulet Iii RJ, Crean CD, Brown ZD, Fruehwald AM, Yang Z, Boswell HS, Nakshatri H and Gabig TG. MOZ and MOZ-CBP cooperate with NF-B to activate transcription from NF-B-dependent promoters. Experimental Hematology. 2007; 35:1782-1792.
78. Cohen I, Poreba E, Kamieniarz K and Schneider R. Histone modifiers in cancer: friends or foes? Genes Cancer. 2011; 2:631-647. doi: 10.1177/1947601911417176.
79. Giannini G, Cabri W, Fattorusso C and Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem. 2012; 4:1439-1460.
80. Martinez-Redondo P and Vaquero A. The diversity of histone versus nonhistone sirtuin substrates. Genes Cancer. 2013; 4:148-163. doi: 10.1177/1947601913483767.
81. Bolden JE, Peart MJ and Johnstone RW. Anticancer activities of histone deacetylase inhibitors
157. Nat Rev Drug Discov. 2006; 5:769-784.
82. Ropero S and Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007; 1:19-25.
83. Minucci S, Nervi C, Lo CF and Pelicci PG. Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene. 2001; 20:3110-3115.
84. Zelent A, Guidez F, Melnick A, Waxman S and Licht JD. Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene. 2001; 20:7186-7203.
85. Cress WD and Seto E. Histone deacetylases, transcriptional control, and cancer. J Cell Physiol. 2000; 184:1-16.
86. Subramanyam D, Belair CD, Barry-Holson KQ, Lin H, Kogan SC, Passegue E and Blelloch R. PML-RAR{alpha} and Dnmt3a1 cooperate in vivo to promote acute promyelocytic leukemia. Cancer Res. 2010; 70:8792-8801.
87. Minucci S and Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006; 6:38-51.
88. Uribesalgo I and Di CL. Dynamics of epigenetic modifications in leukemia. Brief Funct Genomics. 2011; 10:18-29.
89. de TH and Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer. 2010; 10:775-783.
90. Reikvam H, Hatfield KJ, Kittang AO, Hovland R and Bruserud O. Acute myeloid leukemia with the t(8;21) translocation: clinical consequences and biological implications. J Biomed Biotechnol. 2011; 2011:104631.
91. Huang B, Yang XD, Zhou MM, Ozato K and Chen LF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol. 2009; 29:1375-1387.
92. Lamonica JM, Deng W, Kadauke S, Campbell AE, Gamsjaeger R, Wang H, Cheng Y, Billin AN, Hardison RC, Mackay JP and Blobel GA. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc Natl Acad Sci U S A. 2011; 108:E159-E168.
93. Filippakopoulos P and Knapp S. The bromodomain interaction module. FEBS Lett. 2012; 586:2692-2704.
94. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH and Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149:214-231.
95. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011; 478:529-533.
96. Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE and Haldar SM. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013; 154:569-582.
97. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI and Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153:320-334.
98. Zhang W, Prakash C, Sum C, Gong Y, Li Y, Kwok JJ, Thiessen N, Pettersson S, Jones SJ, Knapp S, Yang H and Chin KC. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J Biol Chem. 2012; 287:43137-43155.
99. Garcia JS, Jain N and Godley LA. An update on the safety and efficacy of decitabine in the treatment of myelodysplastic syndromes. Onco Targets Ther. 2010; 3:1-13.
100. Amatori S, Bagaloni I, Donati B and Fanelli M. DNA demethylating antineoplastic strategies: a comparative point of view. Genes Cancer. 2010; 1:197-209. doi: 10.1177/1947601910365081.
101. Estey EH. Epigenetics in clinical practice: the examples of azacitidine and decitabine in myelodysplasia and acute myeloid leukemia. Leukemia. 2013; 27:1803-1812.
102. Joeckel TE and Lubbert M. Clinical results with the DNA hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in patients with myelodysplastic syndromes: an update. Semin Hematol. 2012; 49:330-341.
103. Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS, Cortes J and Kantarjian HM. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004; 103:1635-1640.
104. Silverman LR, McKenzie DR, Peterson BL, Holland JF, Backstrom JT, Beach CL and Larson RA. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006; 24:3895-3903.
105. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009; 10:223-232.
106. Font P. Azacitidine for the treatment of patients with acute myeloid leukemia with 20%-30% blasts and multilineage dysplasia. Adv Ther. 2011; 28 Suppl 3:1-9.
107. Dombret H and Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2015.
108. Malik P and Cashen AF. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag Res. 2014; 6:53-61.
109. Voso MT, Lo-Coco F and Fianchi L. Epigenetic therapy of myelodysplastic syndromes and acute myeloid leukemia. Curr Opin Oncol. 2015; 27:532-539.
110. Griffiths EA and Gore SD. Epigenetic therapies in MDS and AML. Adv Exp Med Biol. 2013; 754:253-283.
111. Ruter B, Wijermans PW and Lubbert M. Superiority of prolonged low-dose azanucleoside administration? Results of 5-aza-2’-deoxycytidine retreatment in high-risk myelodysplasia patients. Cancer. 2006; 106:1744-1750.
112. Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F, Estrov Z, Ferrajoli A, Wierda W, Shan J, Davis J, Giles F, Saba HI, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia.
88. Blood. 2007; 109:52-57.
113. Cashen AF, Schiller GJ, O’Donnell MR and DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010; 28:556-561.
114. Chuang JC, Warner SL, Vollmer D, Vankayalapati H, Redkar S, Bearss DJ, Qiu X, Yoo CB and Jones PA. S110, a 5-Aza-2’-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol Cancer Ther. 2010; 9:1443-1450.
115. Coral S, Parisi G, Nicolay HJ, Colizzi F, Danielli R, Fratta E, Covre A, Taverna P, Sigalotti L and Maio M. Immunomodulatory activity of SGI-110, a 5-aza-2’-deoxycytidine-containing demethylating dinucleotide. Cancer Immunol Immunother. 2013; 62:605-614.
116. Foulks JM, Parnell KM, Nix RN, Chau S, Swierczek K, Saunders M, Wright K, Hendrickson TF, Ho KK, McCullar MV and Kanner SB. Epigenetic drug discovery: targeting DNA methyltransferases. J Biomol Screen. 2012; 17:2-17.
117. Lavelle D, Saunthararajah Y, Vaitkus K, Singh M, Banzon V, Phiasivongsva P, Redkar S, Kanekal S, Bearss D, Shi C, Inloes R and DeSimone J. S110, a novel decitabine dinucleotide, increases fetal hemoglobin levels in baboons (P. anubis). J Transl Med. 2010; 8:92.
118. Montalban-Bravo G and Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia. 2015; 29:760-769.
119. Blum KA, Liu Z, Lucas DM, Chen P, Xie Z, Baiocchi R, Benson DM, Devine SM, Jones J, Andritsos L, Flynn J, Plass C, Marcucci G, Chan KK, Grever MR and Byrd JC. Phase I trial of low dose decitabine targeting DNA hypermethylation in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: dose-limiting myelosuppression without evidence of DNA hypomethylation. Br J Haematol. 2010; 150:189-195.
120. Stathis A, Hotte SJ, Chen EX, Hirte HW, Oza AM, Moretto P, Webster S, Laughlin A, Stayner LA, McGill S, Wang L, Zhang WJ, Espinoza-Delgado I, Holleran JL, Egorin MJ and Siu LL. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin Cancer Res. 2011; 17:1582-1590.
121. Stewart DJ, Issa JP, Kurzrock R, Nunez MI, Jelinek J, Hong D, Oki Y, Guo Z, Gupta S and Wistuba II. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res. 2009; 15:3881-3888.
122. Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013; 340:622-626.
123. Stein EM. Molecular Pathways: IDH2 Mutations-Co-opting Cellular Metabolism for Malignant Transformation. Clin Cancer Res. 2016; 22:16-19.
124. Fredly H, Gjertsen BT and Bruserud O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: the effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin Epigenetics. 2013; 5:12.
125. Kuendgen A, Strupp C, Aivado M, Bernhardt A, Hildebrandt B, Haas R, Germing U and Gattermann N. Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid. Blood. 2004; 104:1266-1269.
126. Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes J, O’Brien S, Estey E, et al. Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006; 108:3271-3279.
127. Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A, Pierce S, Estey EH, Issa JP, Kantarjian HM and Garcia-Manero G. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007; 110:2302-2308.
128. Raffoux E, Cras A, Recher C, Boelle PY, de LA, Turlure P, Marolleau JP, Reman O, Gardin C, Victor M, Maury S, Rousselot P, Malfuson JV, Maarek O, Daniel MT, Fenaux P, et al. Phase 2 clinical trial of 5-azacitidine, valproic acid, and all-trans retinoic acid in patients with high-risk acute myeloid leukemia or myelodysplastic syndrome. Oncotarget. 2010; 1:34-42. doi: 10.18632/oncotarget.106
129. Craddock C, Quek L, Goardon N, Freeman S, Siddique S, Raghavan M, Aztberger A, Schuh A, Grimwade D, Ivey A, Virgo P, Hills R, McSkeane T, Arrazi J, Knapper S, Brookes C, et al. Azacitidine fails to eradicate leukemic stem/progenitor cell populations in patients with acute myeloid leukemia and myelodysplasia. Leukemia. 2013; 27:1028-1036.
130. Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM and Frankel SR. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007; 109:31-39.
131. Kirschbaum M, Frankel P, Popplewell L, Zain J, Delioukina M, Pullarkat V, Matsuoka D, Pulone B, Rotter AJ, Espinoza-Delgado I, Nademanee A, Forman SJ, Gandara D and Newman E. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2011; 29:1198-1203.
132. Silva G, Cardoso BA, Belo H and Almeida AM. Vorinostat induces apoptosis and differentiation in myeloid malignancies: genetic and molecular mechanisms. PLoS One. 2013; 8:e53766.
133. Zhang C, Richon V, Ni X, Talpur R and Duvic M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J Invest Dermatol. 2005; 125:1045-1052.
134. Zhao WL, Wang L, Liu YH, Yan JS, Leboeuf C, Liu YY, Wu WL, Janin A, Chen Z and Chen SJ. Combined effects of histone deacetylase inhibitor and rituximab on non-Hodgkin’s B-lymphoma cells apoptosis. Exp Hematol. 2007; 35:1801-1811.
135. Chen R, Frankel P, Popplewell L, Siddiqi T, Ruel N, Rotter A, Thomas SH, Mott M, Nathwani N, Htut M, Nademanee A, Forman SJ and Kirschbaum M. A phase II study of vorinostat and rituximab for treatment of newly diagnosed and relapsed/refractory indolent non-Hodgkin lymphoma. Haematologica. 2015; 100:357-362.
136. Duvic M and Dimopoulos M. The safety profile of vorinostat (suberoylanilide hydroxamic acid) in hematologic malignancies: A review of clinical studies. Cancer Treatment Reviews. 2016; 43:58-66.
137. Laubach JP, Moreau P, San-Miguel JF and Richardson PG. Panobinostat for the Treatment of Multiple Myeloma. Clin Cancer Res. 2015; 21:4767-4773.
138. Scuto A, Kirschbaum M, Kowolik C, Kretzner L, Juhasz A, Atadja P, Pullarkat V, Bhatia R, Forman S, Yen Y and Jove R. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood. 2008; 111:5093-5100.
139. Rosato R, Hock S, Dent P, Dai Y and Grant S. LBH-589 (panobinostat) potentiates fludarabine anti-leukemic activity through a JNK- and XIAP-dependent mechanism. Leuk Res. 2012; 36:491-498.
140. Xie C, Drenberg C, Edwards H, Caldwell JT, Chen W, Inaba H, Xu X, Buck SA, Taub JW, Baker SD and Ge Y. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PLoS One. 2013; 8:e79106.
141. Tan P, Wei A, Mithraprabhu S, Cummings N, Liu HB, Perugini M, Reed K, Avery S, Patil S, Walker P, Mollee P, Grigg A, D’Andrea R, Dear A and Spencer A. Dual epigenetic targeting with panobinostat and azacitidine in acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood Cancer J. 2014; 4:e170.
142. O’Connor OA, Horwitz S, Masszi T, Van HA, Brown P, Doorduijn J, Hess G, Jurczak W, Knoblauch P, Chawla S, Bhat G, Choi MR, Walewski J, Savage K, Foss F, Allen LF, et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol. 2015; 33:2492-2499.
143. Bodiford A, Bodge M, Talbott MS and Reddy NM. Profile of belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Onco Targets Ther. 2014; 7:1971-1977.
144. Sawas A, Radeski D and O’Connor OA. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: a perspective review. Ther Adv Hematol. 2015; 6:202-208.
145. Kirschbaum MH, Foon KA, Frankel P, Ruel C, Pulone B, Tuscano JM and Newman EM. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California Cancer Consortium Study. Leuk Lymphoma. 2014; 55:2301-2304.
146. Cashen A, Juckett M, Jumonville A, Litzow M, Flynn PJ, Eckardt J, LaPlant B, Laumann K, Erlichman C and DiPersio J. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Ann Hematol. 2012; 91:33-38.
147. Kalac M, Scotto L, Marchi E, Amengual J, Seshan VE, Bhagat G, Ulahannan N, Leshchenko VV, Temkin AM, Parekh S, Tycko B and O’Connor OA. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011; 118:5506-5516.
148. Bose P and Grant S. Orphan drug designation for pracinostat, volasertib and alvocidib in AML. Leuk Res. 2014; 38:862-865.
149. Yoo CB and Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006; 5:37-50.
150. Prebet T, Sun Z, Figueroa ME, Ketterling R, Melnick A, Greenberg PL, Herman J, Juckett M, Smith MR, Malick L, Paietta E, Czader M, Litzow M, Gabrilove J, Erba HP, Gore SD, et al. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: results of the US Leukemia Intergroup trial E1905. J Clin Oncol. 2014; 32:1242-1248.
151. Jona A, Khaskhely N, Buglio D, Shafer JA, Derenzini E, Bollard CM, Medeiros LJ, Illes A, Ji Y and Younes A. The histone deacetylase inhibitor entinostat (SNDX-275) induces apoptosis in Hodgkin lymphoma cells and synergizes with Bcl-2 family inhibitors. Exp Hematol. 2011; 39:1007-1017.
152. Frys S, Simons Z, Hu Q, Barth MJ, Gu JJ, Mavis C, Skitzki J, Song L, Czuczman MS and Hernandez-Ilizaliturri FJ. Entinostat, a novel histone deacetylase inhibitor is active in B-cell lymphoma and enhances the anti-tumour activity of rituximab and chemotherapy agents. Br J Haematol. 2015; 169:506-519.
153. Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ, Reeder C, Joske D, Figg WD, Gardner ER, Steinberg SM, Jaffe ES, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009; 27:5410-5417.
154. Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Borchmann P, Morschhauser F, Wilhelm M, Pinter-Brown L, Padmanabhan S, Shustov A, Nichols J, Carroll S, Balser J, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012; 30:631-636.
155. Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Morschhauser F, Wilhelm M, Pinter-Brown L, Padmanabhan IS, Shustov A, Nielsen T, Nichols J, Wolfson J, Balser B, et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: pivotal study update demonstrates durable responses. J Hematol Oncol. 2014; 7:11.
156. Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, Panageas K, Wright JJ, Pandolfi PP and Nimer SD. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008; 14:826-832.
157. Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, Lucas DM, Fischer B, Shank R, Tejaswi SL, Binkley P, Wright J, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005; 105:959-967.
158. Valdez BC, Brammer JE, Li Y, Murray D, Liu Y, Hosing C, Nieto Y, Champlin RE and Andersson BS. Romidepsin targets multiple survival signaling pathways in malignant T cells. Blood Cancer J. 2015; 5:e357.
159. Ghoneum M and Gollapudi S. Synergistic apoptotic effect of arabinoxylan rice bran (MGN-3/Biobran) and curcumin (turmeric) on human multiple myeloma cell line U266 in vitro. Neoplasma. 2011; 58:118-123.
160. Nebbioso A, Carafa V, Benedetti R and Altucci L. Trials with ‘epigenetic’ drugs: an update. Mol Oncol. 2012; 6:657-682.
161. Aggarwal BB, Deb L and Prasad S. Curcumin differs from tetrahydrocurcumin for molecular targets, signaling pathways and cellular responses. Molecules. 2015; 20:185-205.
162. Eliseeva ED, Valkov V, Jung M and Jung MO. Characterization of novel inhibitors of histone acetyltransferases. Mol Cancer Ther. 2007; 6:2391-2398.
163. Hlubek F, Lohberg C, Meiler J, Jung A, Kirchner T and Brabletz T. Tip60 is a cell-type-specific transcriptional regulator. J Biochem. 2001; 129:635-641.
164. Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, Balusu R, Koul S, Atadja P, Marquez VE and Bhalla KN. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009; 114:2733-2743.
165. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della PA, III, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012; 492:108-112.
166. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012; 8:890-896.
167. Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011; 20:53-65.
168. Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, Waters NJ, Chesworth R, Moyer MP, Copeland RA, Richon VM and Pollock RM. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013; 122:1017-1025.
169. Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010; 468:1119-1123.
170. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, et al. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067-1073.
171. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146:904-917.
172. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L and Sims RJ, III. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011; 108:16669-16674.
173. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011; 478:524-528.
174. Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE and Weinstock DM. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012; 120:2843-2852.