



RNA based individualized drug selection in breast cancer patients without patient-matched normal tissue
2020-01-10
While standard RNA expression tests stratify patients into risk groups, RNA-Seq can guide personalized drug selection based on expressed mutations, fusion genes, and differential expression between tumor and normal tissue. This research identified breast cancer related and drug related genes that are expressed uniformly across our normal samples.
Normal expression from formalin and frozen healthy breast tissue samples using Roche Kapa Ribo Erase and Illumina Tru Seq RNA Access, and fat tissue. Tumor DE using 10 formalin total RNA tumor samples and 1 frozen targeted RNA tumor sample.
Dr. Michael Forster from the Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany said, "Breast cancer is the most common cancer affecting women, with over 265,000 newly diagnosed cases in the USA and over 70,000 in Germany, respectively."
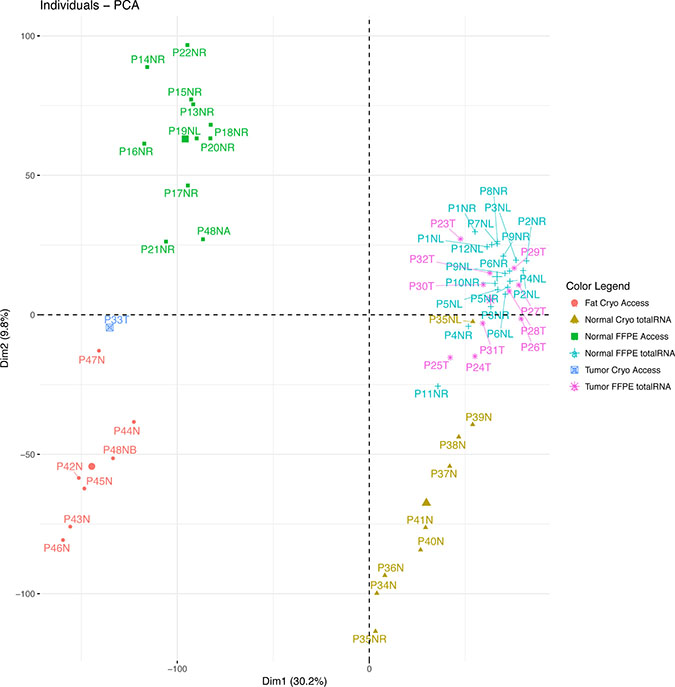
Figure 2: Principal component analysis based on gene expression. The plot shows four distinct clusters of samples: The sequencing methods segregate on the first dimension. The adipocyte content or asservation methods segregate on the second dimension, where the pure adipocyte samples including the fat-rich sample half P48NB segregate in the lower left quadrant and the adipocyte-rich breast tissue samples in the lower right quadrant. The low-fat samples segregate on the upper half, including the fat-free sample half P48NA. Tumor segregation is not distinct.
Even when a patient-matched sample of normal breast tissue is available, additional healthy samples are required to distinguish normal gene expression variability in healthy breast tissue from pathological gene expression in tumor tissue. To address these problems and allow RNA based differential expression analysis to be carried out for breast cancer patients with or without patient-matched healthy breast tissue, we collected healthy normal ductal tissue from breast reduction operations and from resected healthy tissue that was adjacent to tumor tissue. The research team therefore generated healthy normal breast ductal tissue reference expression data from formalin fixed paraffin embedded samples as well as from fresh frozen samples. For quality control, they performed extensive differential expression analyses as summarized in Figure 1, within each group of healthy samples, between groups of healthy samples, between patient-matched pairs of left and right healthy breast tissue, and between tumor and healthy samples.
Their reference data will allow personalized differential expression and immune repertoire analyses to be performed for breast cancer patients who have no remaining healthy breast tissue, with the aim of matching the differential expression data to suitable drugs.
The Michael Forster research team concluded, "Our gene expression and immune repertoire reference data aim to help identify true aberrations and ultimately guide experimental breast cancer drug selection in last-line patients with or without patient-matched normal tissue."
Full text - https://doi.org/10.18632/oncotarget.25981
Correspondence to - Michael Forster - [email protected]
Keywords - breast cancer sequencing, normal RNA expression, drug selection, formalin fixed paraffin embedded, fresh frozen
Copyright © 2026 Rapamycin Press LLC dba Impact Journals
Oncotarget ® is a registered trademark of Rapamycin Press LLC
Impact Journals ® is a registered trademark of Rapamycin Press LLC
RAPAMYCIN PRESS ® is a registered trademark of Rapamycin Press LLC