



Oncotarget: AKT1(E17K) mutation induces mammary hyperplasia but prevents HER2-driven tumorigenesis
2020-12-15
Oncotarget published "Oncogenic AKT1(E17K) mutation induces mammary hyperplasia but prevents HER2-driven tumorigenesis" which reported that one such genetic lesion is the somatic AKT1 mutation, which has been identified in 4-8% of breast cancer patients.
To determine how this mutation contributes to mammary tumorigenesis, the authors constructed a genetically engineered mouse model that conditionally expresses human AKT1 in the mammary epithelium.
Although AKT1 is only weakly constitutively active and does not promote proliferation in vitro, it is capable of escaping negative feedback inhibition to exhibit sustained signaling dynamics in vitro.
Consistently, both virgin and multiparous AKT1 mice develop mammary gland hyperplasia that do not progress to carcinoma.
Moreover, AKT1 prevents HER2-driven mammary tumor formation, in part through negative feedback inhibition of RTK signaling. Analysis of TCGA breast cancer data revealed that the mRNA expression, total protein levels, and phosphorylation of various RTKs are decreased in human tumors harboring AKT1.
Dr. Alex Toker from Harvard Medical School said, "One of the most frequently deregulated pathways in human cancers is the phosphoinositide 3-kinase (PI 3-K) and Akt signaling cascade."
Other breast cancer mutations prevalent in this pathway include mutational or epigenetic inactivation of Phosphatase and Tensin Homolog, a lipid phosphatase that terminates PI 3-K signaling by dephosphorylating PIP3. Both oncogenic PIK3CA mutations and inactivation of PTEN lead to excessive and constitutive activation of Akt and downstream signaling, resulting in uncontrolled proliferation and increased cellular survival .
In the context of breast cancer, the AKT1 somatic mutation was first identified in breast cancer but is also found in lung, bladder, endometrial, urothelial and prostate cancers.
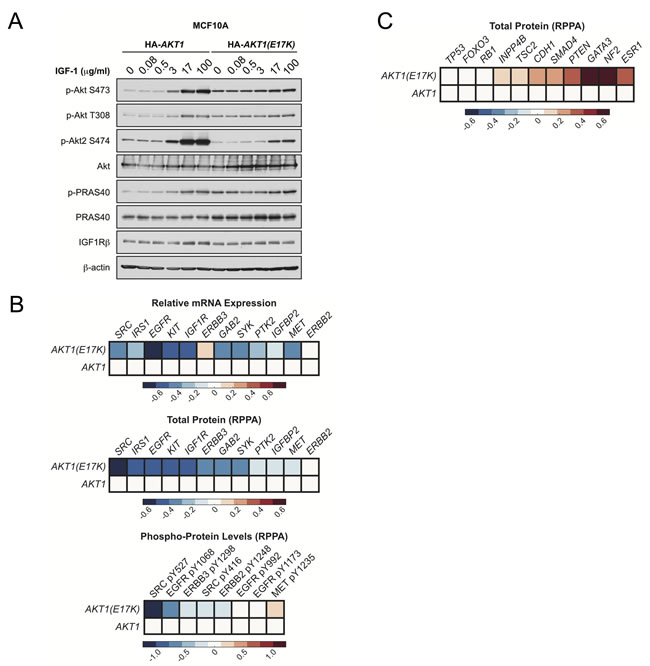
Figure 7: AKT1(E17K) suppresses RTK expression and phosphorylation through negative feedback inhibition. A. MCF10A cells expressing tet-on HA-AKT1/pTRIPZ or HA-AKT1(E17K)/pTRIPZ were treated with 150 ng/ml doxycycline for 48 h to induce AKT1 or AKT1(E17K) expression. Cells were serum-starved for 16 h and then treated with the indicated concentrations of IGF-1 for 10 min. Whole cell lysates were subjected to immunoblotting. B.-C. Data for mRNA, RPPA total protein and RPPA phospho-protein was downloaded from the TCGA data set on the cBIO Portal (http://www.cbioportal.org/public-portal/). Heat maps were generated using MatLab and represent microarray/RPPA values for the indicated genes/proteins, which are expressed relative to values in the wild-type AKT1 group.
Interestingly, in breast cancer AKT1 is mutually exclusive with PIK3CA and PTEN mutations, although in other cancers such as endometrial carcinoma, these mutations frequently co-exist in the same tumor.
Similarly, knock-in of the AKT1 mutation into MCF-7 ER-positive cells in which oncogenic PIK3CA has been restored to the wild-type allele restores proliferation and tumor growth in vivo, arguing that at least in ER-positive luminal breast cancer cells, AKT1 can function as a bona fide oncogene.
To date, no studies have examined the capacity of AKT1 to drive mammary cancer in a genetically engineered mouse model. Previous studies have addressed the contribution of AKT1 activity to mammary tumorigenesis using constitutively active AKT1 transgenes driven by the mouse mammary tumor virus promoter.
The Toker Research Team concluded in their Oncotarget Priority Research Paper that the findings presented in this study have implications for therapeutic intervention and development of targeted therapies for both RTKs as well as PI 3-K and Akt.
There are presently numerous phase I and II clinical trials with a variety of allosteric and catalytic Akt small molecule inhibitors for therapeutic benefit in breast and other solid tumors.
Since inhibition of Akt using catalytic inhibitors leads to the up-regulation of RTK signaling, and AKT1 tumors show suppressed levels and phosphorylation of RTKs, one implication is that breast cancer patients with HER2 amplification would not benefit from treatment with Akt inhibitors.
Instead, combination therapy with Akt inhibitors followed by RTK inhibition may be a more effective therapeutic strategy.
Whether the same mechanism of RTK suppression and escape of feedback inhibition applies to these mutants remains to be determined.
Sign up for free Altmetric alerts about this article
DOI - https://doi.org/10.18632/oncotarget.8191
Full text - https://www.oncotarget.com/article/8191/text/
Correspondence to - Alex Toker - [email protected]
Keywords - Akt, breast cancer, PI 3-kinase, HER2, estrogen receptor
About Oncotarget
Oncotarget is a biweekly, peer-reviewed, open access biomedical journal covering research on all aspects of oncology.
To learn more about Oncotarget, please visit https://www.oncotarget.com or connect with:
SoundCloud - https://soundcloud.com/oncotarget
Facebook - https://www.facebook.com/Oncotarget/
Twitter - https://twitter.com/oncotarget
LinkedIn - https://www.linkedin.com/company/oncotarget
Pinterest - https://www.pinterest.com/oncotarget/
Reddit - https://www.reddit.com/user/Oncotarget/
Oncotarget is published by Impact Journals, LLC please visit http://www.ImpactJournals.com or connect with @ImpactJrnls
Media Contact
[email protected]
18009220957x105
Copyright © 2026 Rapamycin Press LLC dba Impact Journals
Oncotarget ® is a registered trademark of Rapamycin Press LLC
Impact Journals ® is a registered trademark of Rapamycin Press LLC
RAPAMYCIN PRESS ® is a registered trademark of Rapamycin Press LLC