The cover for issue 6 of Oncotarget features Figure 5, "AKT-specific inhibitor MK2206 decreases cell viability, spheroid formation, and migration of SETD2 deficient ccRCC-derived cells," by Terzo, et al.
Upregulation of the PI3K pathway has been implicated in the initiation and progression of several types of cancer, including renal cell carcinoma (RCC). Although several targeted therapies have been developed for RCC, durable and complete responses are exceptional. Thus, advanced RCC remains a lethal disease, underscoring the need of robust biomarker-based strategies to treat RCC. We report a synthetic lethal interaction between inhibition of phosphatidylinositol 3-kinase beta (PI3Kβ) and loss of SETD2 methyltransferase. Clear cell RCC (ccRCC)-derived SETD2 knockout 786-0 and SETD2 mutant A498 cells treated with TGX221 (PI3Kβ-specific) and AZD8186 (PI3Kβ- and δ-specific) inhibitors displayed decreased cell viability, cell growth, and migration compared to SETD2 proficient 786-0 cells. Inhibition of the p110 δ and α isoforms alone had modest (δ) and no (α) effect on ccRCC cell viability, growth, and migration. In vivo, treatment of SETD2 mutant A498 cells, but not SETD2 proficient 786-0 cells, with AZD8186 significantly decreased tumor growth. Interestingly, inhibition of the downstream effector AKT (MK2206) recapitulated the effects observed in AZD8186-treated SETD2 deficient cells. Our data show that specific inhibition of PI3Kβ causes synthetic lethality with SETD2 loss and suggest targeting of the AKT downstream effector pathway offers a rationale for further translational and clinical investigation of PI3Kβ-specific inhibitors in ccRCC.
"Clear cell renal cell carcinoma, the most common histological subtype, accounts for the majority of RCC-related deaths."
The diversity of molecular pathways requiring SETD2s methylating activity underscores the enzymes crucial role in maintaining cellular homeostasis and warrants further investigation into molecular networks involving SETD2 that drive cc RCC oncogenesis.
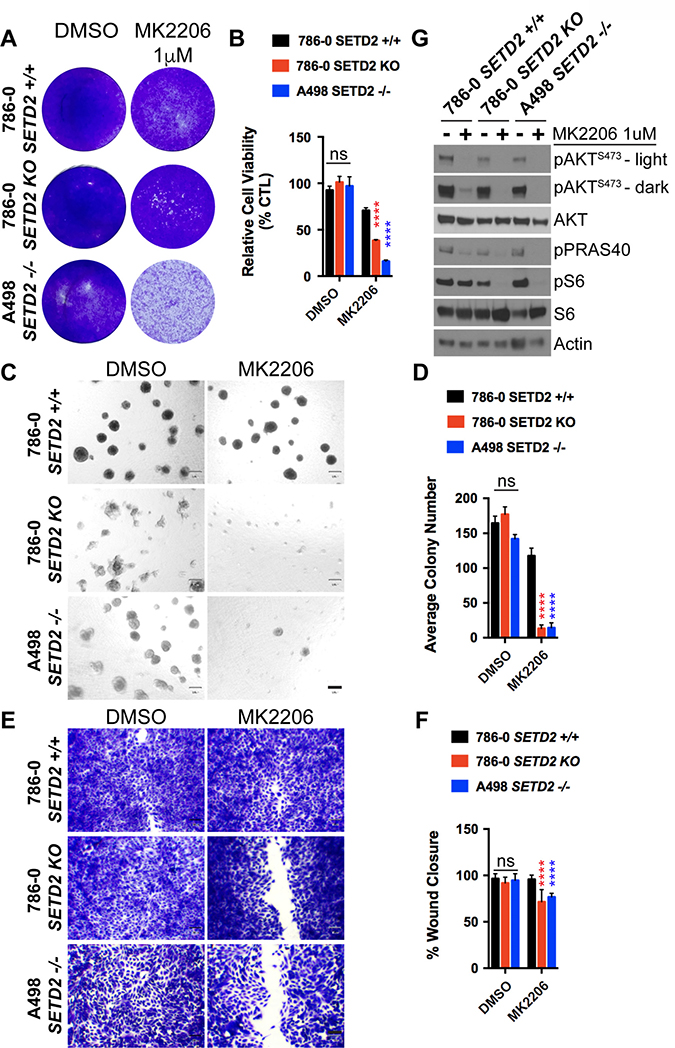
Figure 5: AKT-specific inhibitor MK2206 decreases cell viability, spheroid formation, and migration of SETD2 deficient ccRCC-derived cells. (A) Bright-field microscopy images showing living cells (attached to bottom of well) stained with 0.3% crystal violet solution of SETD2 (+/+) 786-0 and SETD2 (KO) 786-0 and (–/–) A498 cells were treated with vehicle (DMSO) or 1 μM AKT-specific inhibitor MK2206 for 10 days. (B) Bar graph showing relative cell viability as a percentage of CTL of SETD2 (+/+) 786-0 and SETD2 (KO) 786-0 and (–/–) A498 cells in response to treatment with MK2206. ****P < 0.0001; ns, no statistical significance observed. Standard deviations were calculated and represented for all conditions. (C) Phase-contrast pictures showing spheroid formation of SETD2 (+/+) 786-0 and SETD2 (KO) 786-0 and (–/–) A498 cells in response to treatment with vehicle (DMSO) and 500 nM inhibitor for 14 days grown in Matrigel. (D) Bar graph showing average colony number of SETD2 (+/+) 786-0 and SETD2 (KO) 786-0 and (–/–) A498 cells treated with MK2206 inhibitor. ****P < 0.0001; ns, no statistical significance observed. Standard deviations were calculated and represented for all conditions. (E) Bright-field microscopy images showing living cells stained with 0.3% crystal violet solution. (F) Bar graph showing percentage (%) of wound closure compared to cells treated with vehicle (DMSO). (G) Western blot analysis of indicated proteins showing variations in phosphorylation levels in response to chemical inhibition with MK2206 (AKT-specific). Whole-cell protein lysates from cells treated with 1 μM inhibitor for 24 hours were resolved by SDS-PAGE. Actin is a loading control.
A recent study utilizing the Genomics of Drug Sensitivity in Cancer database identified that RCC cells with mutated VHL or SETD2 were sensitive to the small molecule PIK3 inhibitor TGX221.
TGX221 was also shown to target cancer cells with CDKN2A and PTEN mutations, suggesting nonspecific inhibition at the molar concentration used in the study.
In this study, we sought to expand on this reported sensitivity by examining the effects of genetic and pharmacologic inhibition of the PI3K-AKT axis and its downstream effectors in more well-defined and in vivo model systems.
We show that SETD2 deficient 786-0 and A498 cells are significantly more sensitive to PI3K -specific and PI3K / -specific inhibitors than SETD2 proficient isogenic paired 786-0 cells, as evidenced by impaired viability, cell migration, spheroid formation, as well as genotype-selective reduced growth in vivo.
Lastly, SETD2 deficient cell lines treated with MK2206 recapitulated the effects observed in AZD8186-treated SETD2 deficient cells, implicating canonical PI3K signaling via AKT as a key mechanism of viability.
"They also demonstrated that TGX221-treated VHL and SETD2 deficient cc RCC-derived cell lines have a significantly reduced migrating and invading capacity when compared to VHL deficient cc RCC-derived cell lines, suggesting a novel molecular connection between PI3K and SETD2.
Our combined data demonstrating that SETD2 deficient cc RCC-derived cells are significantly sensitive, less migratory, and less capable of forming spheroids and that tumor formation of SETD2 mutant xenografts is abrogated, further underscore the biological relevance of this molecular connection between SETD2 and PI3K."
Full text - https://doi.org/10.18632/oncotarget.26567
Correspondence to - W. Kimryn Rathmell - [email protected]
Keywords - SETD2, PI3Kβ, synthetic lethality, renal cell carcinoma


